

Enhanced Vaccine Immunogenicity Enabled by Targeted Cytosolic Delivery of Tumor Antigens into Dendritic Cells
- PMID: 37780364
- PMCID: PMC10540291
- DOI: 10.1021/acscentsci.3c00625
Enhanced Vaccine Immunogenicity Enabled by Targeted Cytosolic Delivery of Tumor Antigens into Dendritic Cells
Abstract
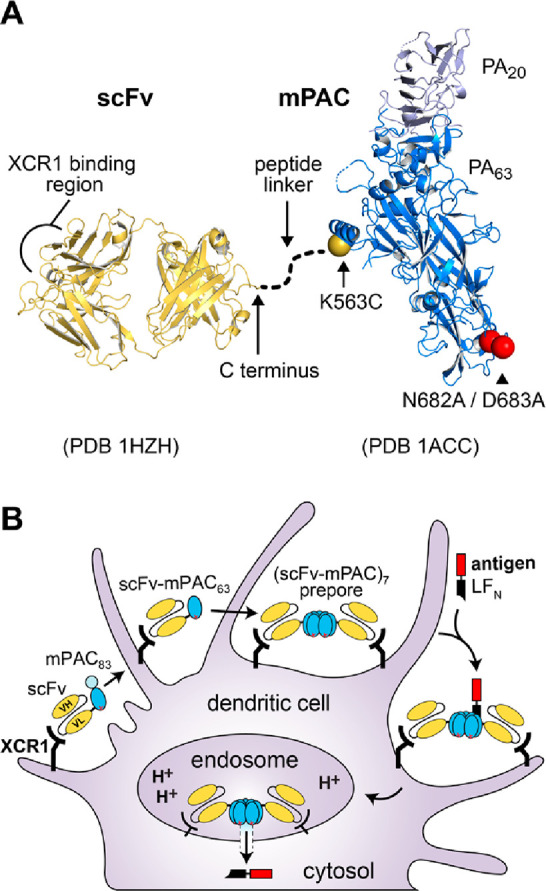
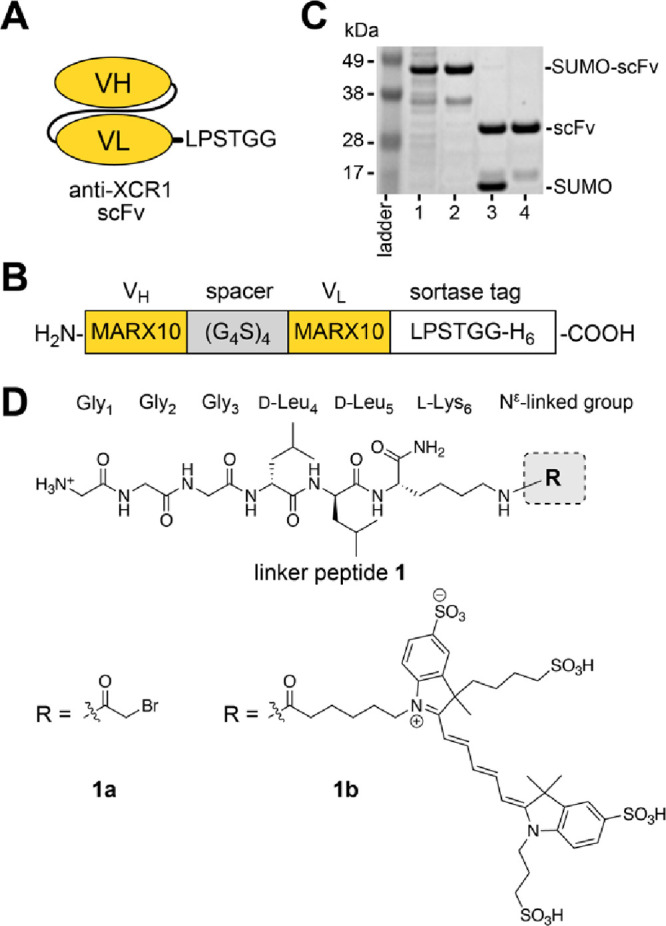
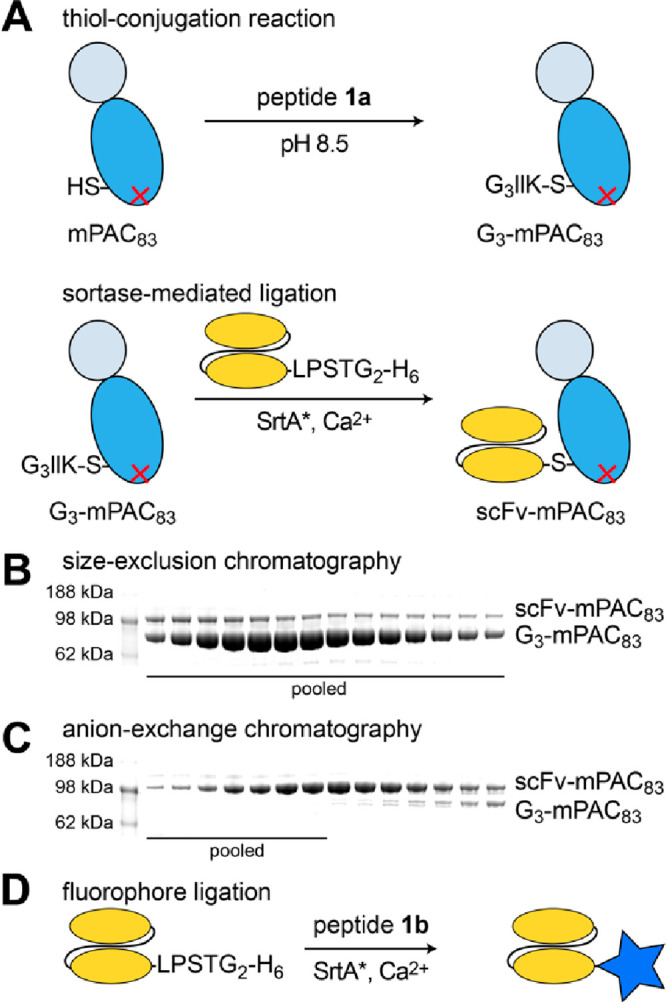
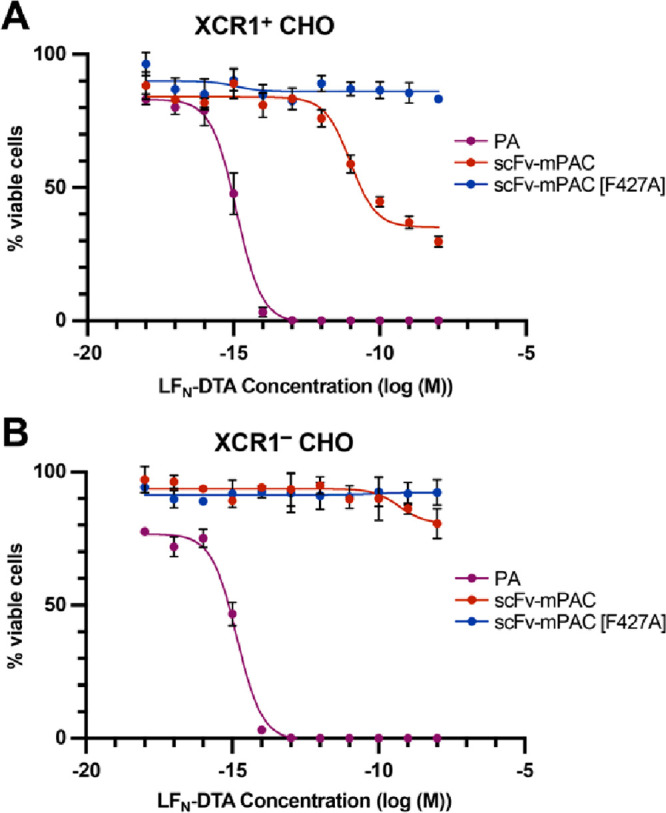
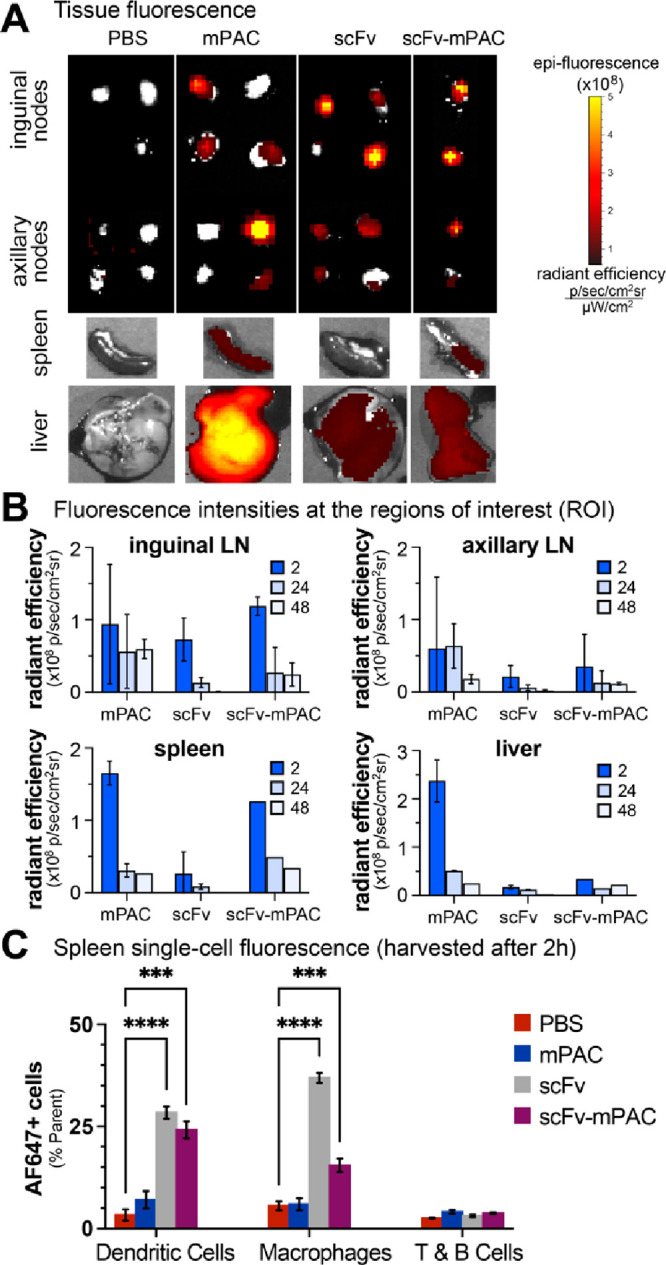
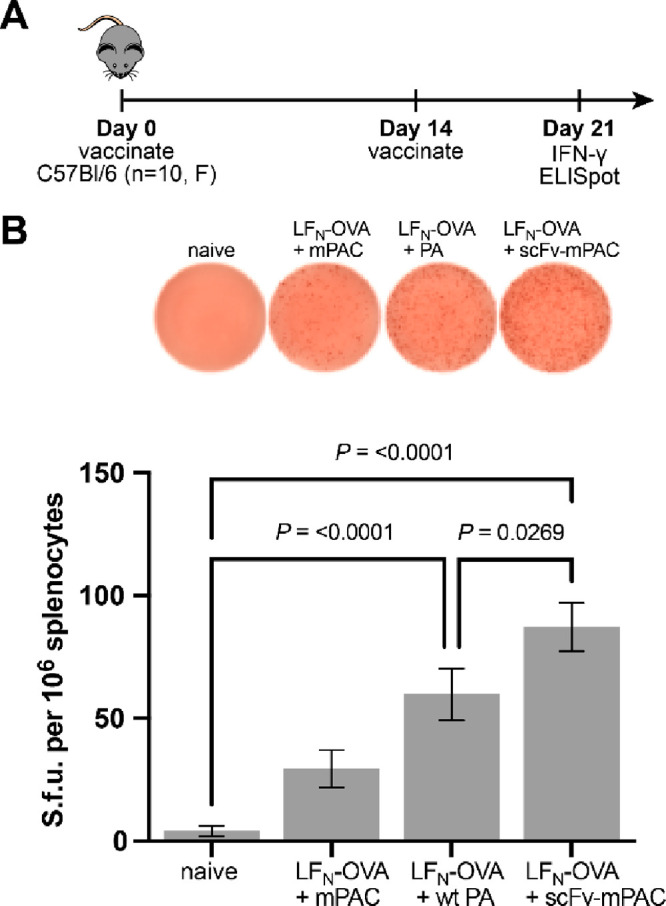
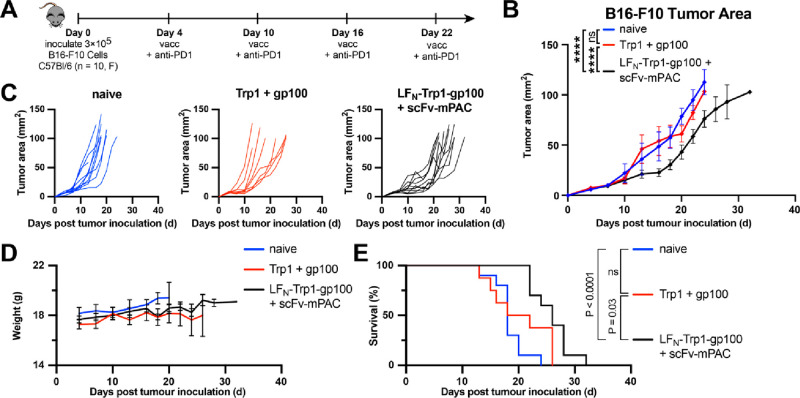
Molecular vaccines comprising antigen peptides and inflammatory cues make up a class of therapeutics that promote immunity against cancer and pathogenic diseases but often exhibit limited efficacy. Here, we engineered an antigen peptide delivery system to enhance vaccine efficacy by targeting dendritic cells and mediating cytosolic delivery. The delivery system consists of the nontoxic anthrax protein, protective antigen (PA), and a single-chain variable fragment (scFv) that recognizes the XCR1 receptor on dendritic cells (DCs). Combining these proteins enabled selective delivery of the N-terminus of lethal factor (LFN) into XCR1-positive cross-presenting DCs. Incorporating immunogenic epitope sequences into LFN showed selective protein translocation in vitro and enhanced the priming of antigen-specific T cells in vivo. Administering DC-targeted constructs with tumor antigens (Trp1/gp100) into mice bearing aggressive B16-F10 melanomas improved mouse outcomes when compared to free antigen, including suppressed tumor growth up to 58% at 16 days post tumor induction (P < 0.0001) and increased survival (P = 0.03). These studies demonstrate that harnessing DC-targeting anthrax proteins for cytosolic antigen delivery significantly enhances the immunogenicity and antitumor efficacy of cancer vaccines.
© 2023 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare the following competing financial interest(s): B.L.P. is a co-founder and/or member of the scientific advisory board of several companies focusing on the development of protein and peptide therapeutics.
Figures
Similar articles
-
A multi-trimeric fusion of CD40L and gp100 tumor antigen activates dendritic cells and enhances survival in a B16-F10 melanoma DNA vaccine model.Vaccine. 2015 Sep 11;33(38):4798-806. doi: 10.1016/j.vaccine.2015.07.081. Epub 2015 Aug 1. Vaccine. 2015. PMID: 26241951 Free PMC article.
-
Vaccine molecules targeting Xcr1 on cross-presenting DCs induce protective CD8+ T-cell responses against influenza virus.Eur J Immunol. 2015 Feb;45(2):624-35. doi: 10.1002/eji.201445080. Epub 2014 Dec 28. Eur J Immunol. 2015. PMID: 25410055
-
Murine dendritic cells transfected with human GP100 elicit both antigen-specific CD8(+) and CD4(+) T-cell responses and are more effective than DNA vaccines at generating anti-tumor immunity.Int J Cancer. 1999 Nov 12;83(4):532-40. doi: 10.1002/(sici)1097-0215(19991112)83:4<532::aid-ijc16>3.0.co;2-k. Int J Cancer. 1999. PMID: 10508491
-
DC-Based Vaccines for Cancer Immunotherapy.Vaccines (Basel). 2020 Nov 26;8(4):706. doi: 10.3390/vaccines8040706. Vaccines (Basel). 2020. PMID: 33255895 Free PMC article. Review.
-
Idiotypic vaccination for B-cell malignancies as a model for therapeutic cancer vaccines: from prototype protein to second generation vaccines.Haematologica. 2002 Sep;87(9):989-1001. Haematologica. 2002. PMID: 12217812 Review.
Cited by
-
DNA Anchoring Strength Directly Correlates with Spherical Nucleic Acid-Based HPV E7 Cancer Vaccine Potency.Nano Lett. 2024 Jun 26;24(25):7629-7636. doi: 10.1021/acs.nanolett.4c01392. Epub 2024 Jun 14. Nano Lett. 2024. PMID: 38874796 Free PMC article.
-
Forced intracellular degradation of xenoantigens as a modality for cell-based cancer immunotherapy.iScience. 2025 Feb 4;28(3):111957. doi: 10.1016/j.isci.2025.111957. eCollection 2025 Mar 21. iScience. 2025. PMID: 40060894 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
