

Genetic diversification patterns in swine influenza A virus (H1N2) in vaccinated and nonvaccinated animals
- PMID: 37780850
- PMCID: PMC10540852
- DOI: 10.3389/fcimb.2023.1258321
Genetic diversification patterns in swine influenza A virus (H1N2) in vaccinated and nonvaccinated animals
Abstract
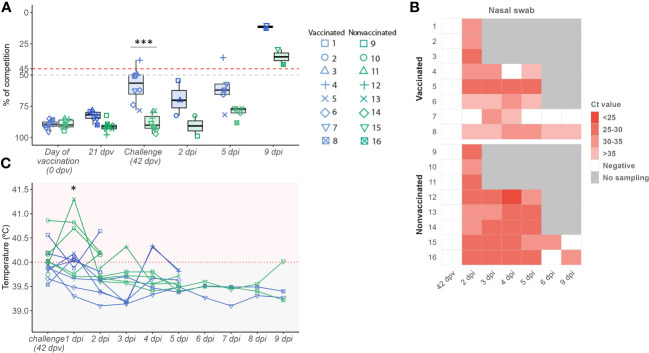
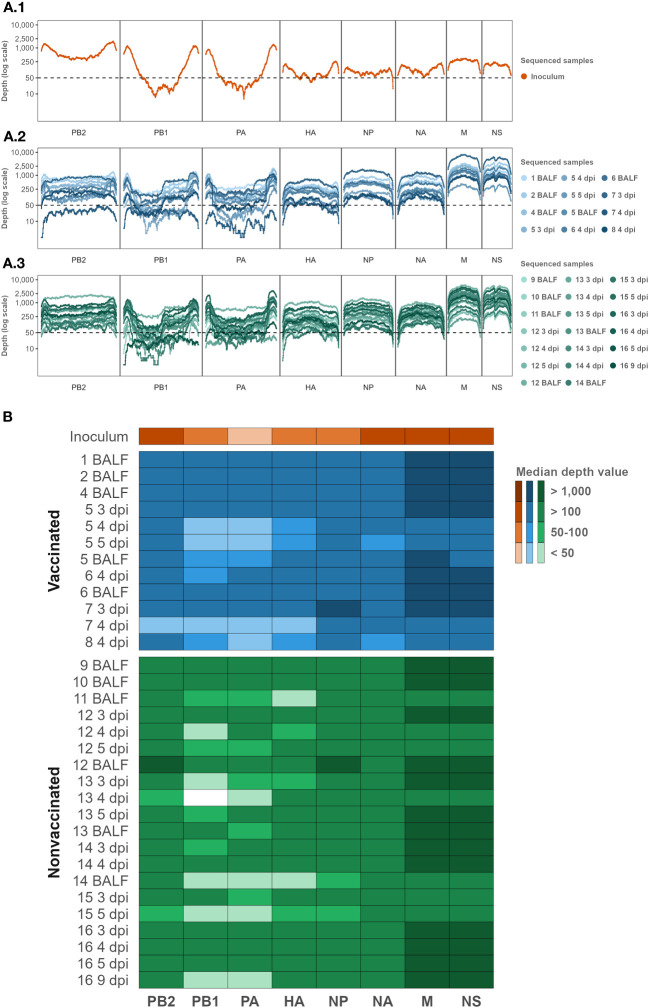
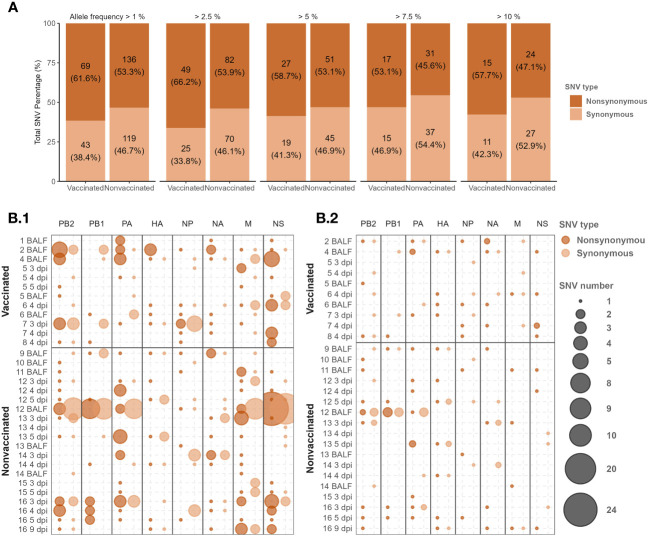
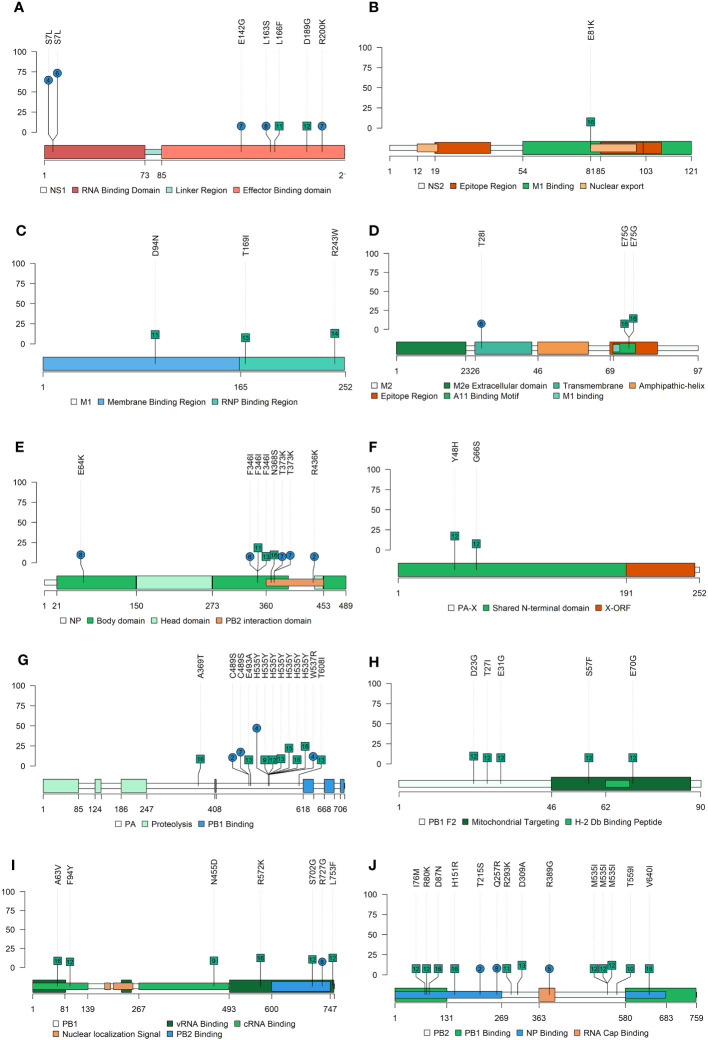
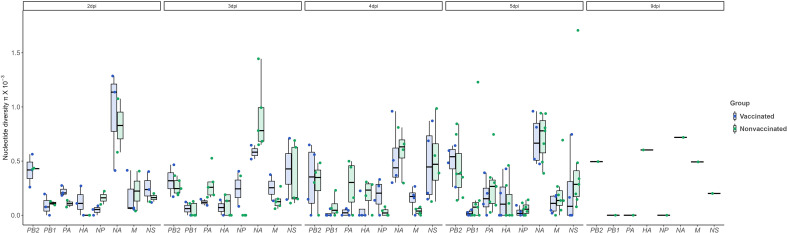
Influenza A viruses (IAVs) are characterized by having a segmented genome, low proofreading polymerases, and a wide host range. Consequently, IAVs are constantly evolving in nature causing a threat to animal and human health. In 2009 a new human pandemic IAV strain arose in Mexico because of a reassortment between two strains previously circulating in pigs; Eurasian "avian-like" (EA) swine H1N1 and "human-like" H1N2, highlighting the importance of swine as adaptation host of avian to human IAVs. Nowadays, although of limited use, a trivalent vaccine, which include in its formulation H1N1, H3N2, and, H1N2 swine IAV (SIAV) subtypes, is one of the most applied strategies to reduce SIAV circulation in farms. Protection provided by vaccines is not complete, allowing virus circulation, potentially favoring viral evolution. The evolutionary dynamics of SIAV quasispecies were studied in samples collected at different times from 8 vaccinated and 8 nonvaccinated pigs, challenged with H1N2 SIAV. In total, 32 SIAV genomes were sequenced by next-generation sequencing, and subsequent variant-calling genomic analysis was carried out. Herein, a total of 364 de novo single nucleotide variants (SNV) were found along all genetic segments in both experimental groups. The nonsynonymous substitutions proportion found was greater in vaccinated animals suggesting that H1N2 SIAV was under positive selection in this scenario. The impact of each substitution with an allele frequency greater than 5% was hypothesized according to previous literature, particularly in the surface glycoproteins hemagglutinin and neuraminidase. The H1N2 SIAV quasispecies evolution capacity was evidenced, observing different evolutionary trends in vaccinated and nonvaccinated animals.
Keywords: NGS; quasispecies; swine influenza virus; vaccination; viral evolution.
Copyright © 2023 López-Valiñas, Valle, Pérez, Darji, Chiapponi, Ganges, Segalés and Núñez.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Vaccination against swine influenza in pigs causes different drift evolutionary patterns upon swine influenza virus experimental infection and reduces the likelihood of genomic reassortments.Front Cell Infect Microbiol. 2023 Mar 13;13:1111143. doi: 10.3389/fcimb.2023.1111143. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 36992684 Free PMC article.
-
Identification and Characterization of Swine Influenza Virus H1N1 Variants Generated in Vaccinated and Nonvaccinated, Challenged Pigs.Viruses. 2021 Oct 16;13(10):2087. doi: 10.3390/v13102087. Viruses. 2021. PMID: 34696517 Free PMC article.
-
[Swine influenza virus: evolution mechanism and epidemic characterization--a review].Wei Sheng Wu Xue Bao. 2009 Sep;49(9):1138-45. Wei Sheng Wu Xue Bao. 2009. PMID: 20030049 Review. Chinese.
-
Genetic and biological characterisation of an avian-like H1N2 swine influenza virus generated by reassortment of circulating avian-like H1N1 and H3N2 subtypes in Denmark.Virol J. 2013 Sep 18;10:290. doi: 10.1186/1743-422X-10-290. Virol J. 2013. PMID: 24047399 Free PMC article.
-
Swine Influenza A Viruses and the Tangled Relationship with Humans.Cold Spring Harb Perspect Med. 2021 Mar 1;11(3):a038737. doi: 10.1101/cshperspect.a038737. Cold Spring Harb Perspect Med. 2021. PMID: 31988203 Free PMC article. Review.
Cited by
-
Evolutionary analysis of Hemagglutinin and neuraminidase gene variation in H1N1 swine influenza virus from vaccine intervention in China.Sci Rep. 2024 Nov 20;14(1):28792. doi: 10.1038/s41598-024-80457-4. Sci Rep. 2024. PMID: 39567587 Free PMC article.
-
IAVCP (Influenza A Virus Consensus and Phylogeny): Automatic Identification of the Genomic Sequence of the Influenza A Virus from High-Throughput Sequencing Data.Viruses. 2024 May 29;16(6):873. doi: 10.3390/v16060873. Viruses. 2024. PMID: 38932165 Free PMC article.
References
-
- Andrews S. (2010) FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed September 1, 2021).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical