

BRCC3-Mediated NLRP3 Deubiquitylation Promotes Inflammasome Activation and Atherosclerosis in Tet2 Clonal Hematopoiesis
- PMID: 37781816
- PMCID: PMC10872582
- DOI: 10.1161/CIRCULATIONAHA.123.065344
BRCC3-Mediated NLRP3 Deubiquitylation Promotes Inflammasome Activation and Atherosclerosis in Tet2 Clonal Hematopoiesis
Abstract
Background: Clonal hematopoiesis (CH) has emerged as an independent risk factor for atherosclerotic cardiovascular disease, with activation of macrophage inflammasomes as a potential underlying mechanism. The NLRP3 (NLR family pyrin domain containing 3) inflammasome has a key role in promoting atherosclerosis in mouse models of Tet2 CH, whereas inhibition of the inflammasome product interleukin-1β appeared to particularly benefit patients with TET2 CH in CANTOS (Cardiovascular Risk Reduction Study [Reduction in Recurrent Major CV Disease Events]). TET2 is an epigenetic modifier that decreases promoter methylation. However, the mechanisms underlying macrophage NLRP3 inflammasome activation in TET2 (Tet methylcytosine dioxygenase 2) deficiency and potential links with epigenetic modifications are poorly understood.
Methods: We used cholesterol-loaded TET2-deficient murine and embryonic stem cell-derived isogenic human macrophages to evaluate mechanisms of NLRP3 inflammasome activation in vitro and hypercholesterolemic Ldlr-/- mice modeling TET2 CH to assess the role of NLRP3 inflammasome activation in atherosclerosis.
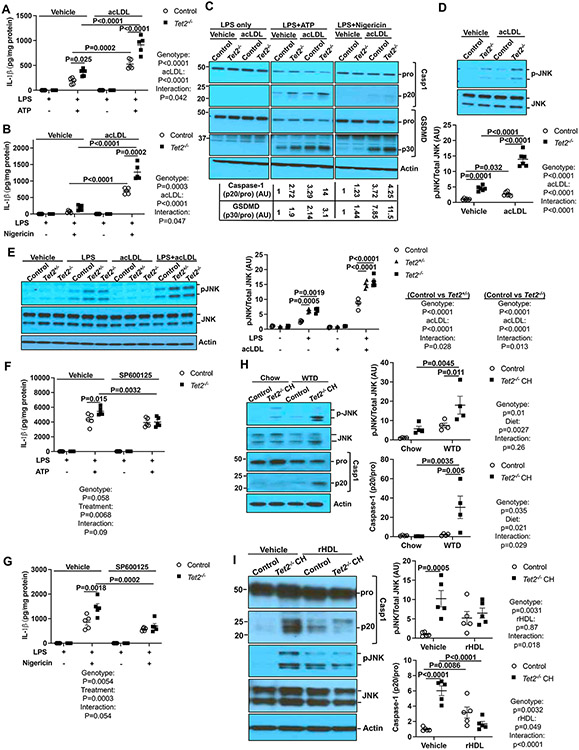
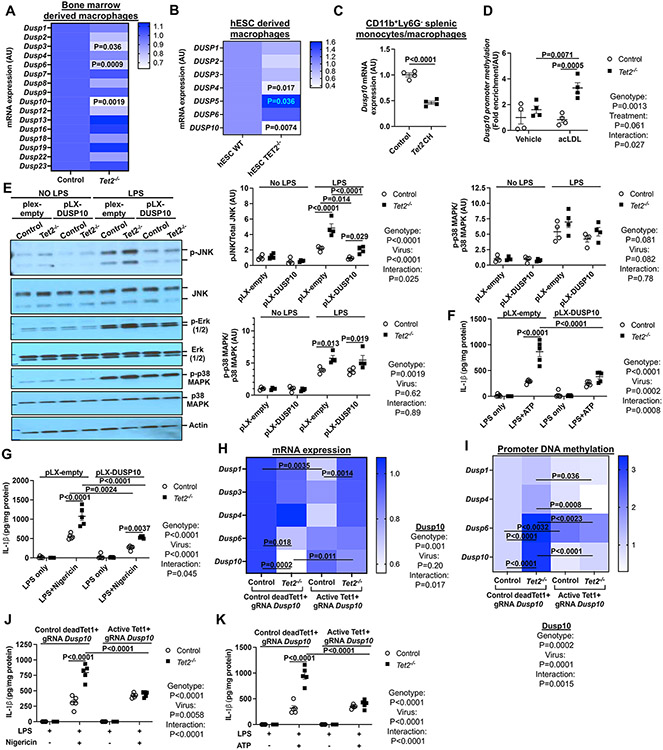
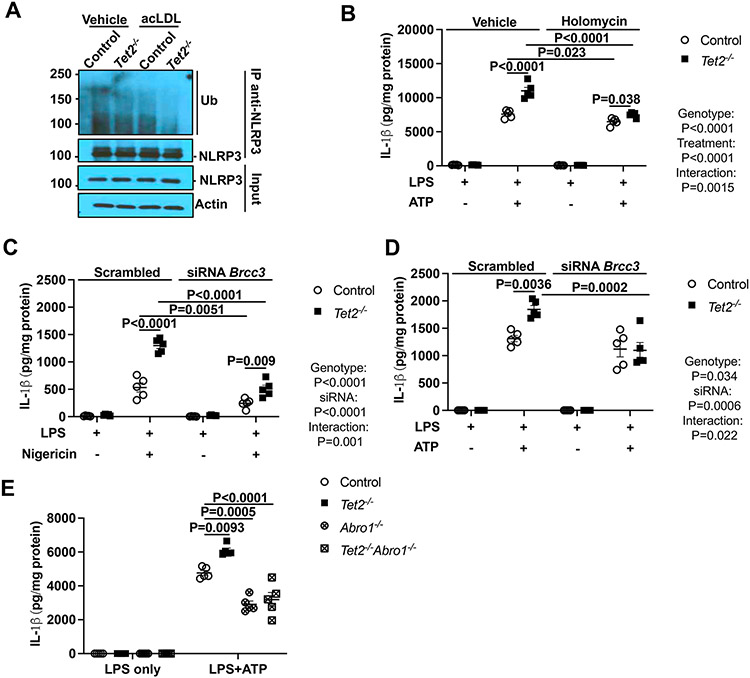
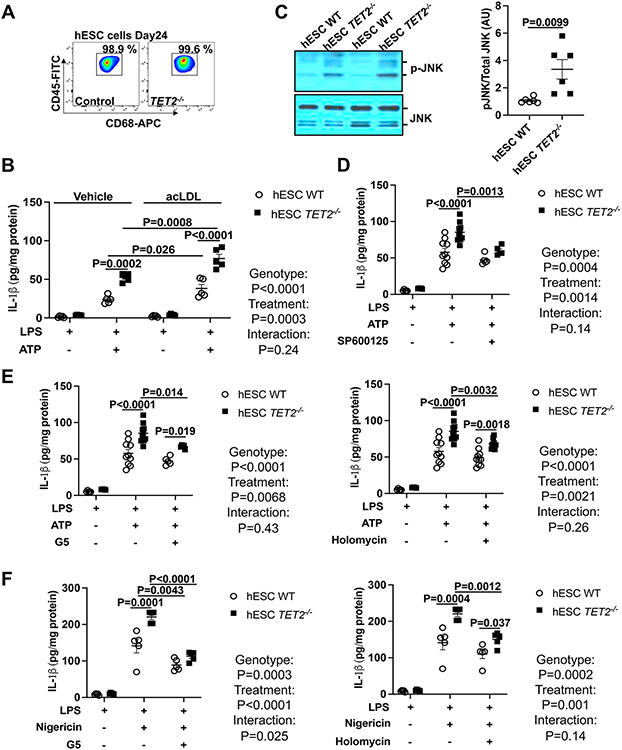
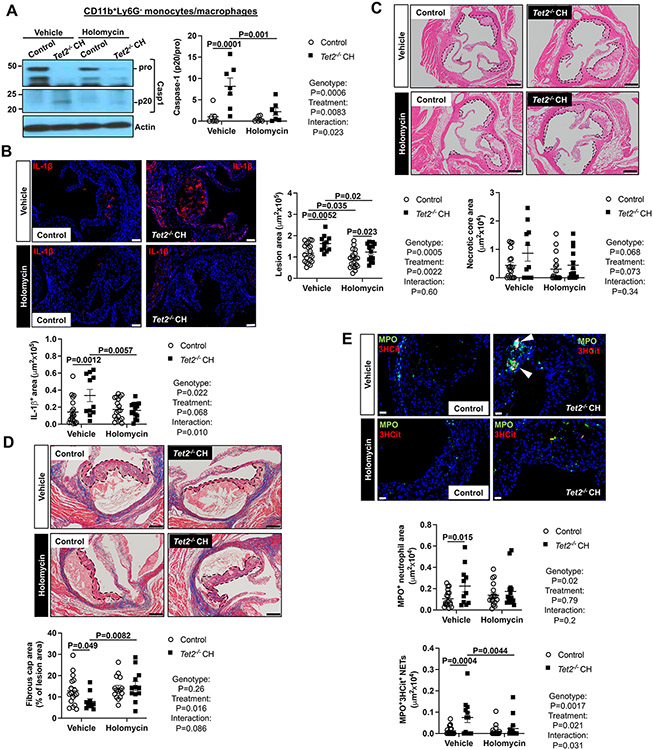
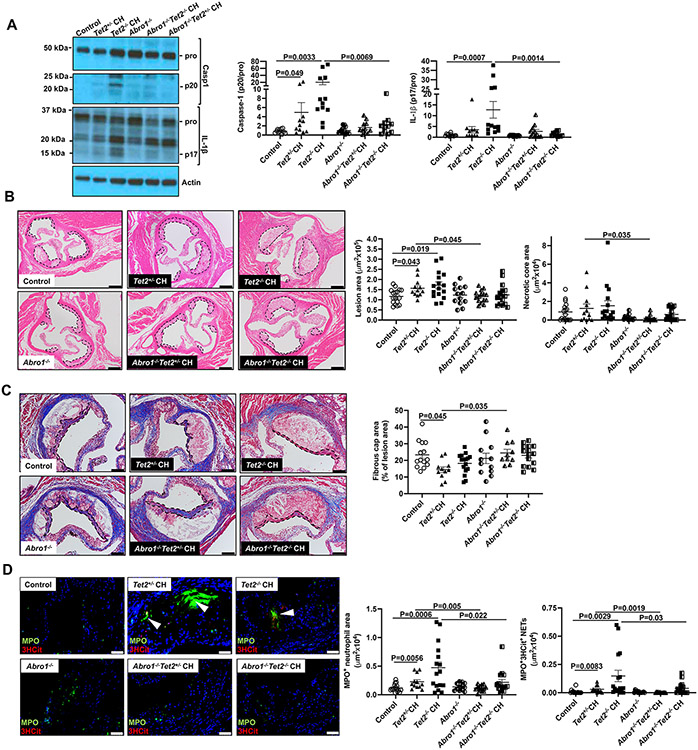
Results: Tet2 deficiency in murine macrophages acted synergistically with cholesterol loading in cell culture and with hypercholesterolemia in vivo to increase JNK1 (c-Jun N-terminal kinase 1) phosphorylation and NLRP3 inflammasome activation. The mechanism of JNK (c-Jun N-terminal kinase) activation in TET2 deficiency was increased promoter methylation and decreased expression of the JNK-inactivating dual-specificity phosphatase Dusp10. Active Tet1-deadCas9-targeted editing of Dusp10 promoter methylation abolished cholesterol-induced inflammasome activation in Tet2-deficient macrophages. Increased JNK1 signaling led to NLRP3 deubiquitylation and activation by the deubiquitinase BRCC3 (BRCA1/BRCA2-containing complex subunit 3). Accelerated atherosclerosis and neutrophil extracellular trap formation (NETosis) in Tet2 CH mice were reversed by holomycin, a BRCC3 deubiquitinase inhibitor, and also by hematopoietic deficiency of Abro1, an essential scaffolding protein in the BRCC3-containing cytosolic complex. Human TET2-/- macrophages displayed increased JNK1 and NLRP3 inflammasome activation, especially after cholesterol loading, with reversal by holomycin treatment, indicating human relevance.
Conclusions: Hypercholesterolemia and TET2 deficiency converge on a common pathway of NLRP3 inflammasome activation mediated by JNK1 activation and BRCC3-mediated NLRP3 deubiquitylation, with potential therapeutic implications for the prevention of cardiovascular disease in TET2 CH.
Keywords: atherosclerosis; clonal hematopoiesis; extracellular traps; hypercholesterolemia; inflammasomes; neutrophils; phosphorylation.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
