

XA4C: eXplainable representation learning via Autoencoders revealing Critical genes
- PMID: 37782668
- PMCID: PMC10569512
- DOI: 10.1371/journal.pcbi.1011476
XA4C: eXplainable representation learning via Autoencoders revealing Critical genes
Abstract
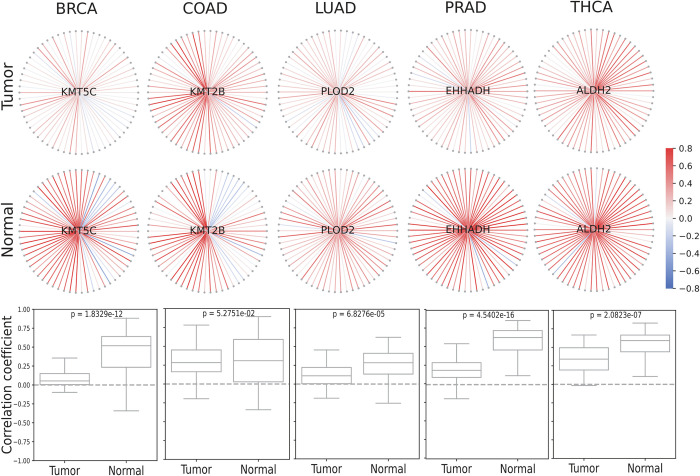
Machine Learning models have been frequently used in transcriptome analyses. Particularly, Representation Learning (RL), e.g., autoencoders, are effective in learning critical representations in noisy data. However, learned representations, e.g., the "latent variables" in an autoencoder, are difficult to interpret, not to mention prioritizing essential genes for functional follow-up. In contrast, in traditional analyses, one may identify important genes such as Differentially Expressed (DiffEx), Differentially Co-Expressed (DiffCoEx), and Hub genes. Intuitively, the complex gene-gene interactions may be beyond the capture of marginal effects (DiffEx) or correlations (DiffCoEx and Hub), indicating the need of powerful RL models. However, the lack of interpretability and individual target genes is an obstacle for RL's broad use in practice. To facilitate interpretable analysis and gene-identification using RL, we propose "Critical genes", defined as genes that contribute highly to learned representations (e.g., latent variables in an autoencoder). As a proof-of-concept, supported by eXplainable Artificial Intelligence (XAI), we implemented eXplainable Autoencoder for Critical genes (XA4C) that quantifies each gene's contribution to latent variables, based on which Critical genes are prioritized. Applying XA4C to gene expression data in six cancers showed that Critical genes capture essential pathways underlying cancers. Remarkably, Critical genes has little overlap with Hub or DiffEx genes, however, has a higher enrichment in a comprehensive disease gene database (DisGeNET) and a cancer-specific database (COSMIC), evidencing its potential to disclose massive unknown biology. As an example, we discovered five Critical genes sitting in the center of Lysine degradation (hsa00310) pathway, displaying distinct interaction patterns in tumor and normal tissues. In conclusion, XA4C facilitates explainable analysis using RL and Critical genes discovered by explainable RL empowers the study of complex interactions.
Copyright: © 2023 Li et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
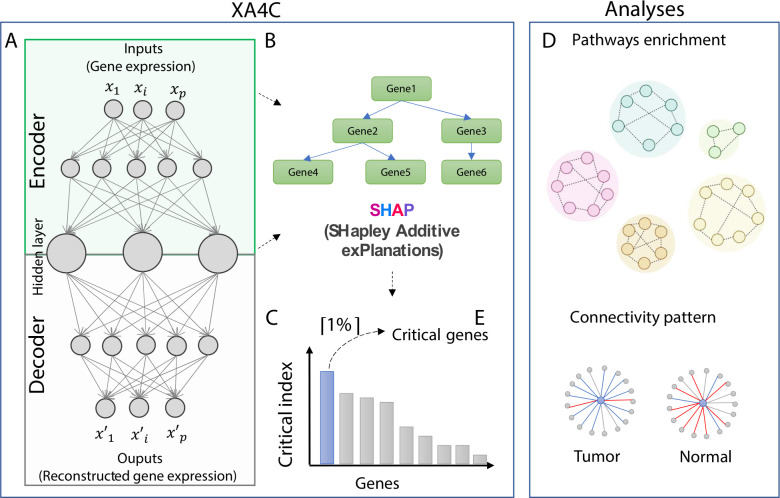
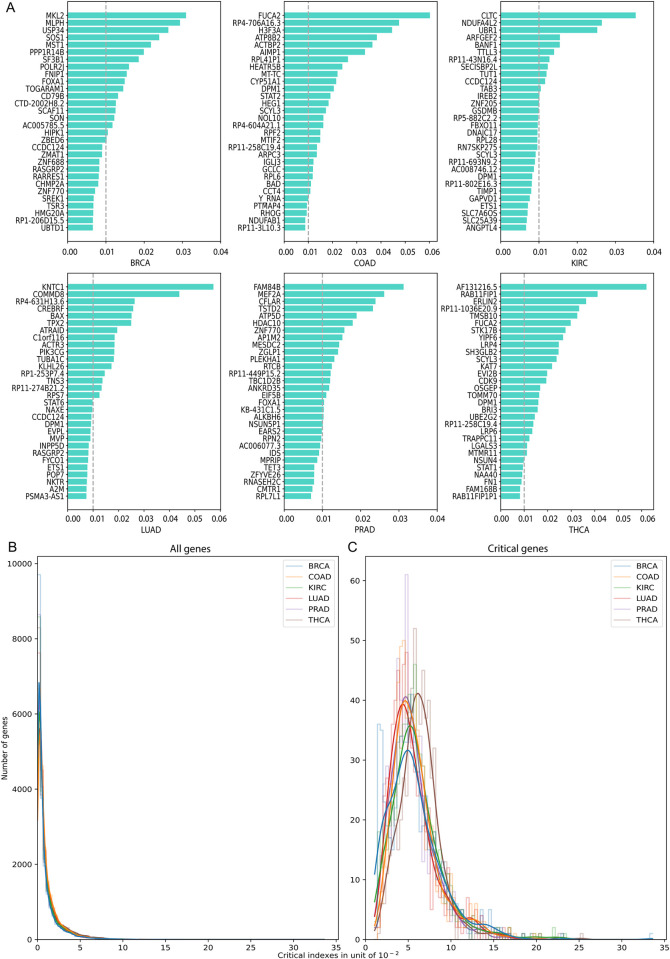
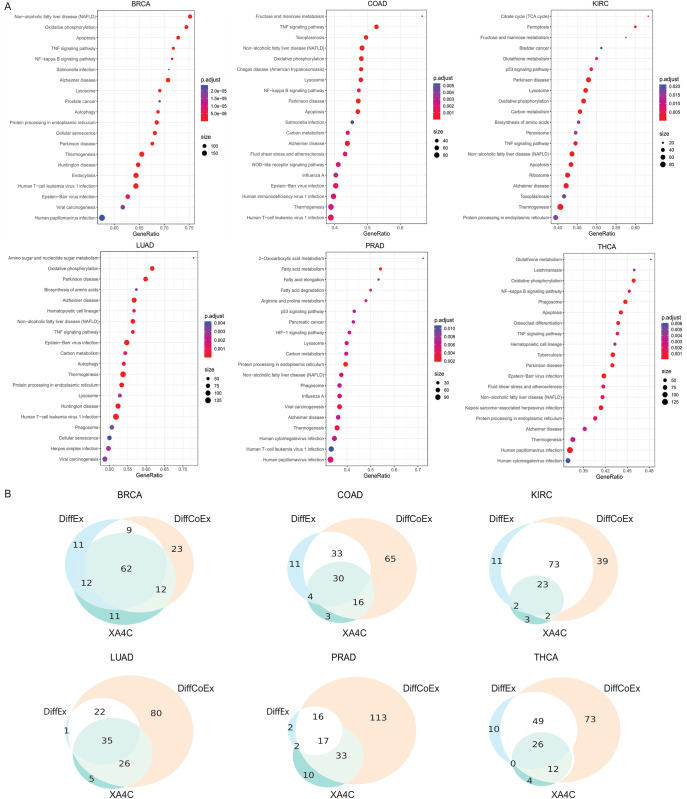
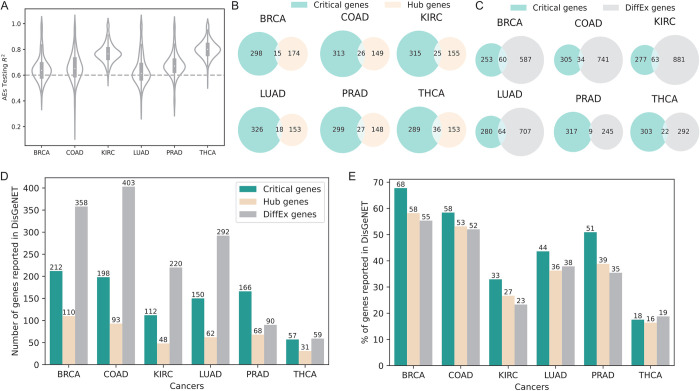
References
-
- Goodfellow I, Bengio Y, Courville A. Deep learning: MIT press; 2016.
-
- Jiayi B, Qing L, Albert L, Guotao Y, Jun Y, Jingjing W, et al.. Autoencoder-transformed transcriptome improves genotype-phenotype association studies. bioRxiv. 2023. 10.1101/2023.07.23.550223. - DOI
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
