

Centuries of genome instability and evolution in soft-shell clam, Mya arenaria, bivalve transmissible neoplasia
- PMID: 37783804
- PMCID: PMC10663159
- DOI: 10.1038/s43018-023-00643-7
Centuries of genome instability and evolution in soft-shell clam, Mya arenaria, bivalve transmissible neoplasia
Abstract
Transmissible cancers are infectious parasitic clones that metastasize to new hosts, living past the death of the founder animal in which the cancer initiated. We investigated the evolutionary history of a cancer lineage that has spread though the soft-shell clam (Mya arenaria) population by assembling a chromosome-scale soft-shell clam reference genome and characterizing somatic mutations in transmissible cancer. We observe high mutation density, widespread copy-number gain, structural rearrangement, loss of heterozygosity, variable telomere lengths, mitochondrial genome expansion and transposable element activity, all indicative of an unstable cancer genome. We also discover a previously unreported mutational signature associated with overexpression of an error-prone polymerase and use this to estimate the lineage to be >200 years old. Our study reveals the ability for an invertebrate cancer lineage to survive for centuries while its genome continues to structurally mutate, likely contributing to the evolution of this lineage as a parasitic cancer.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
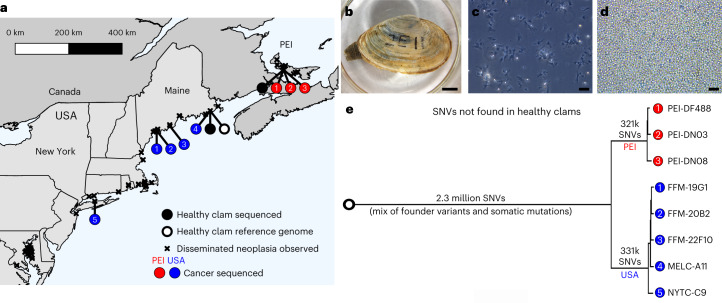


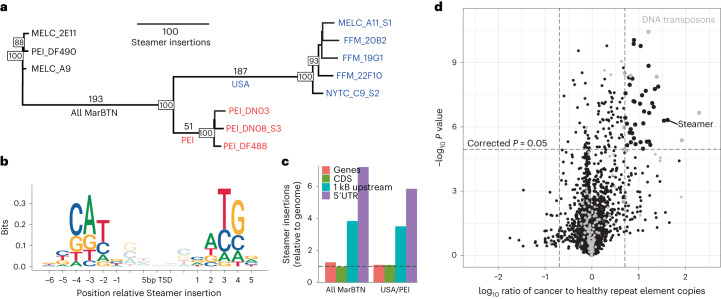











References
-
- Ní Leathlobhair M, Lenski RE. Population genetics of clonally transmissible cancers. Nat. Ecol. Evol. 2022;6:1077–1089. - PubMed
-
- Rebbeck CA, Thomas R, Breen M, Leroi AM, Burt A. Origins and evolution of a transmissible cancer. Evolution. 2009;63:2340–2349. - PubMed
-
- Pearse A-M, Swift K. Transmission of devil facial-tumour disease. Nature. 2006;439:549. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
