

Spatial single-cell mass spectrometry defines zonation of the hepatocyte proteome
- PMID: 37783884
- PMCID: PMC10555842
- DOI: 10.1038/s41592-023-02007-6
Spatial single-cell mass spectrometry defines zonation of the hepatocyte proteome
Abstract
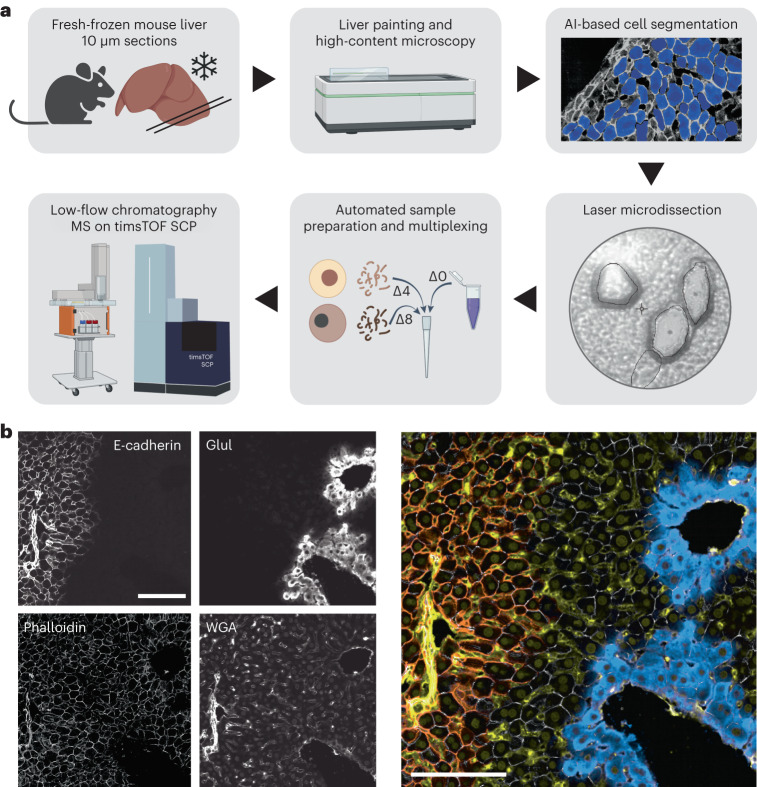
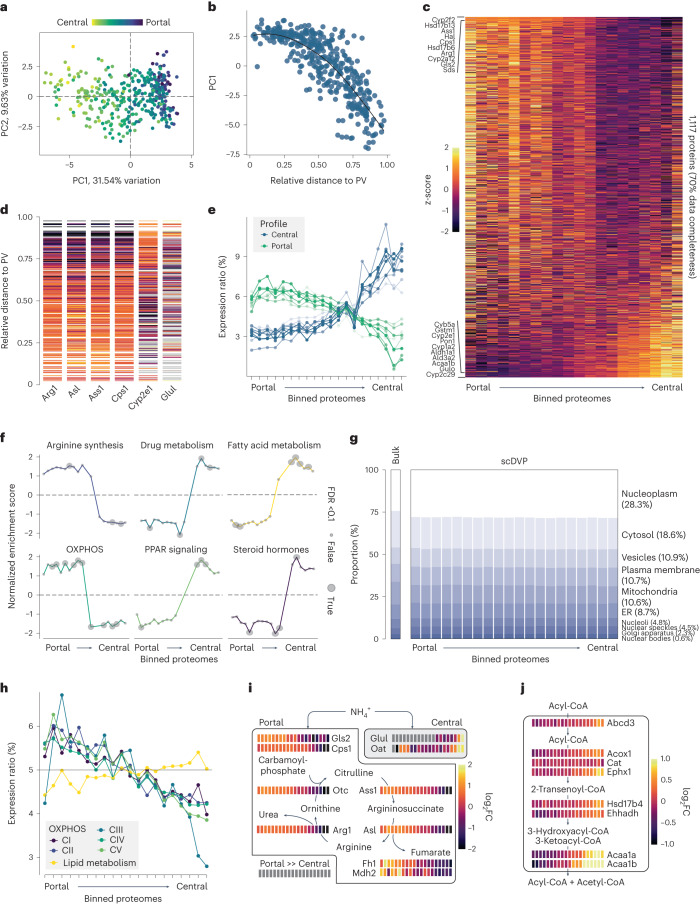
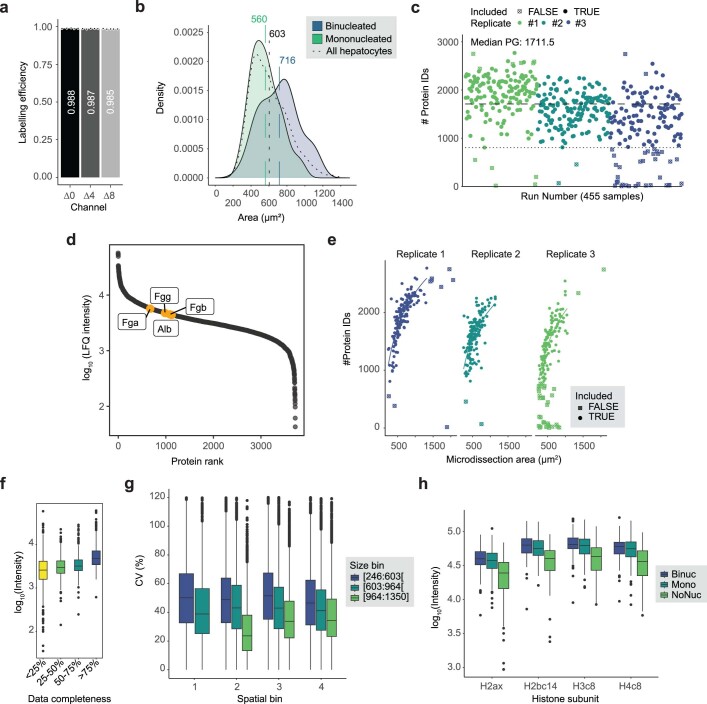
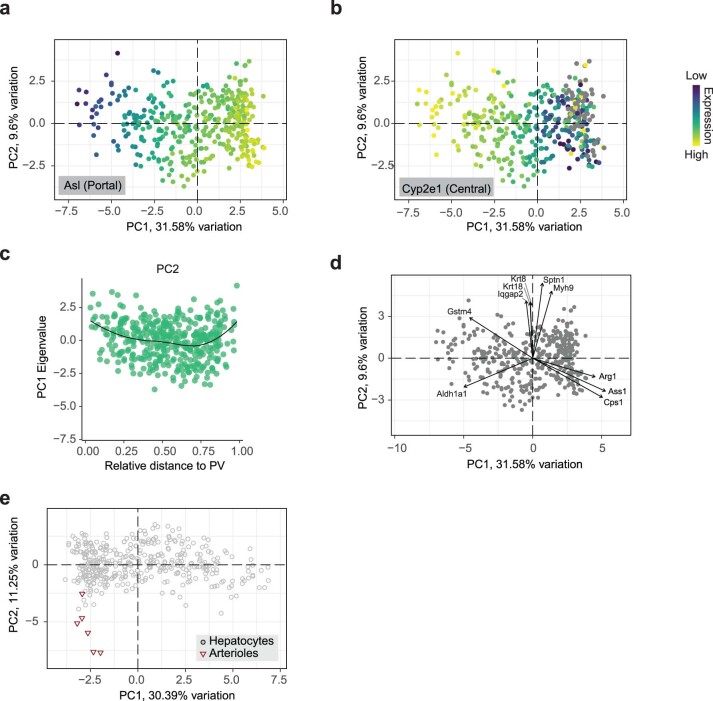
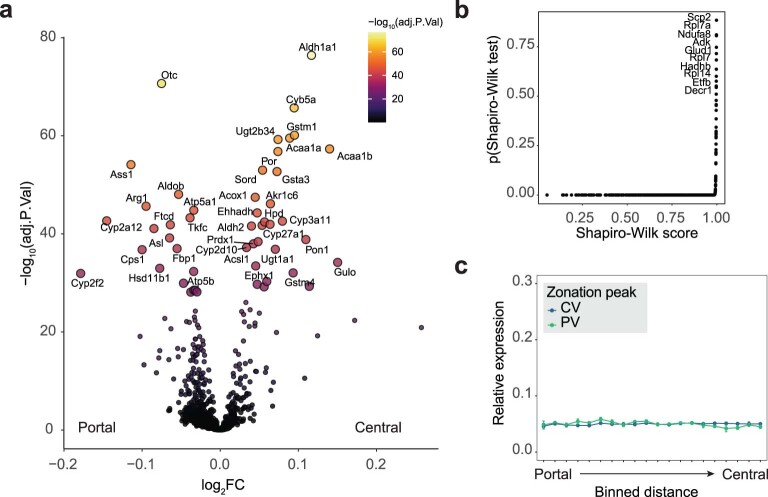
Single-cell proteomics by mass spectrometry is emerging as a powerful and unbiased method for the characterization of biological heterogeneity. So far, it has been limited to cultured cells, whereas an expansion of the method to complex tissues would greatly enhance biological insights. Here we describe single-cell Deep Visual Proteomics (scDVP), a technology that integrates high-content imaging, laser microdissection and multiplexed mass spectrometry. scDVP resolves the context-dependent, spatial proteome of murine hepatocytes at a current depth of 1,700 proteins from a cell slice. Half of the proteome was differentially regulated in a spatial manner, with protein levels changing dramatically in proximity to the central vein. We applied machine learning to proteome classes and images, which subsequently inferred the spatial proteome from imaging data alone. scDVP is applicable to healthy and diseased tissues and complements other spatial proteomics and spatial omics technologies.
© 2023. The Author(s).
Conflict of interest statement
M.M. is an indirect investor in Evosep. All other authors declare no competing interests.
Figures








References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
