

This is a preprint.
Efficient High-Quality Metagenome Assembly from Long Accurate Reads using Minimizer-space de Bruijn Graphs
- PMID: 37786716
- PMCID: PMC10541625
- DOI: 10.1101/2023.07.07.548136
Efficient High-Quality Metagenome Assembly from Long Accurate Reads using Minimizer-space de Bruijn Graphs
Update in
-
High-quality metagenome assembly from long accurate reads with metaMDBG.Nat Biotechnol. 2024 Sep;42(9):1378-1383. doi: 10.1038/s41587-023-01983-6. Epub 2024 Jan 2. Nat Biotechnol. 2024. PMID: 38168989 Free PMC article.
Abstract
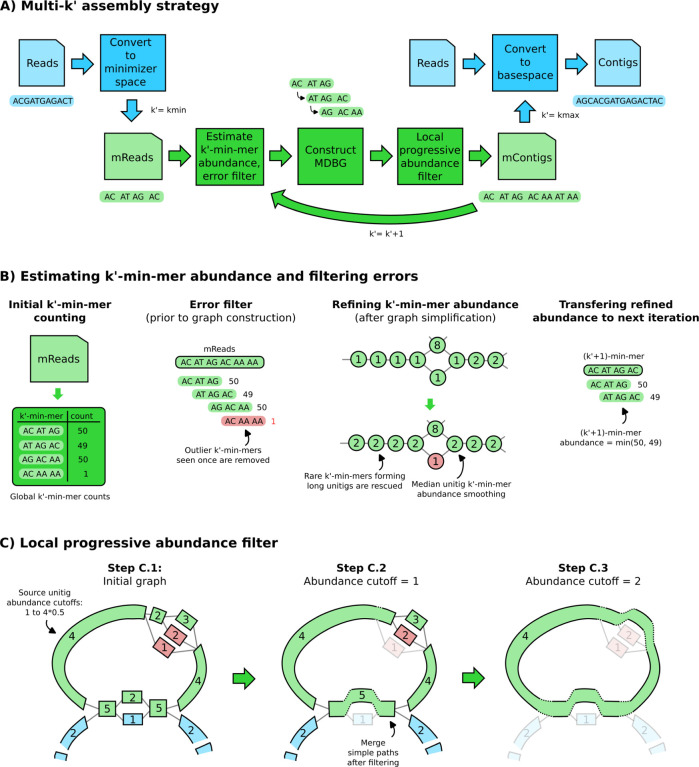
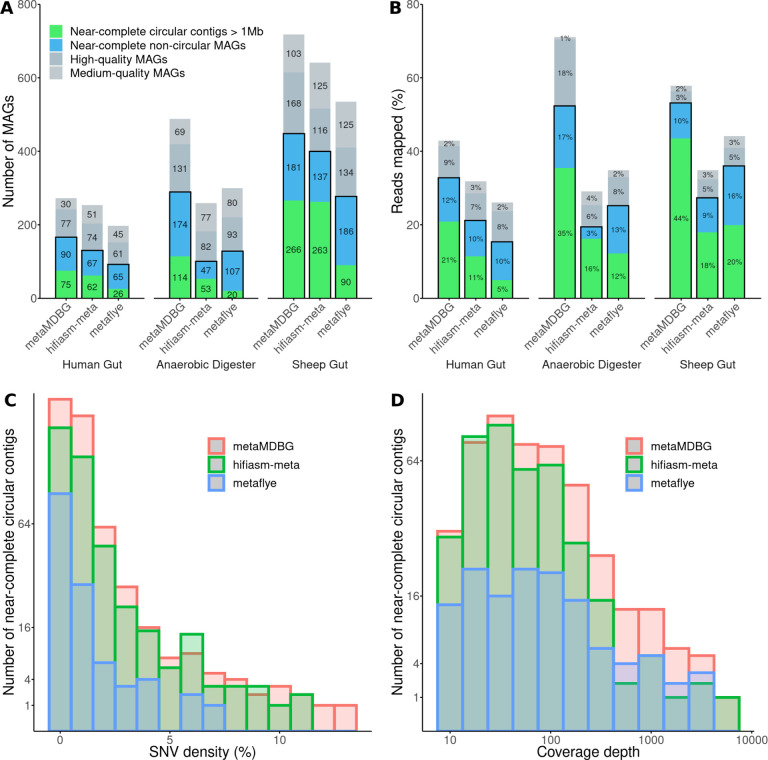
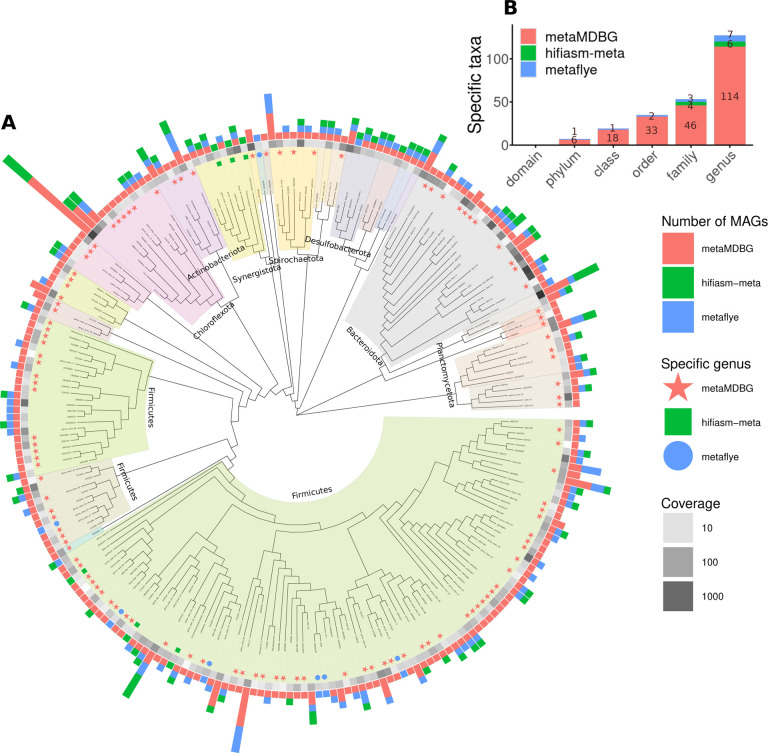
We introduce a novel metagenomics assembler for high-accuracy long reads. Our approach, implemented as metaMDBG, combines highly efficient de Bruijn graph assembly in minimizer space, with both a multi-k' approach for dealing with variations in genome coverage depth and an abundance-based filtering strategy for simplifying strain complexity. The resulting algorithm is more efficient than the state-of-the-art but with better assembly results. metaMDBG was 1.5 to 12 times faster than competing assemblers and requires between one-tenth and one-thirtieth of the memory across a range of data sets. We obtained up to twice as many high-quality circularised prokaryotic metagenome assembled genomes (MAGs) on the most complex communities, and a better recovery of viruses and plasmids. metaMDBG performs particularly well for abundant organisms whilst being robust to the presence of strain diversity. The result is that for the first time it is possible to efficiently reconstruct the majority of complex communities by abundance as near-complete MAGs.
Conflict of interest statement
Competing Interests Statement The authors declare no competing interests.
Figures
References
-
- Quince C., Walker A.W., Simpson J.T., Loman N.J., and Segata N.. Shotgun metagenomics, from sampling to analysis. Nature Biotechnology, 35(9), 2017. - PubMed
-
- Edgar Robert C, Taylor Jeff, Lin Victor, Altman Tomer, Barbera Pierre, Meleshko Dmitry, Lohr Dan, Novakovsky Gherman, Buchfink Benjamin, Al-Shayeb Basem, et al. Petabase-scale sequence alignment catalyses viral discovery. Nature, 602(7895):142–147, 2022. - PubMed
-
- Alneberg J., Bjarnason B.S., De Bruijn I., Schirmer M., Quick J., Ijaz U.Z., Lahti L., Loman N.J., Andersson A.F., and Quince C.. Binning metagenomic contigs by coverage and composition. Nature Methods, 11(11), 2014. - PubMed
Publication types
LinkOut - more resources
Full Text Sources