

Epigenetic balance ensures mechanistic control of MLL amplification and rearrangement
- PMID: 37788669
- PMCID: PMC10591855
- DOI: 10.1016/j.cell.2023.09.009
Epigenetic balance ensures mechanistic control of MLL amplification and rearrangement
Abstract
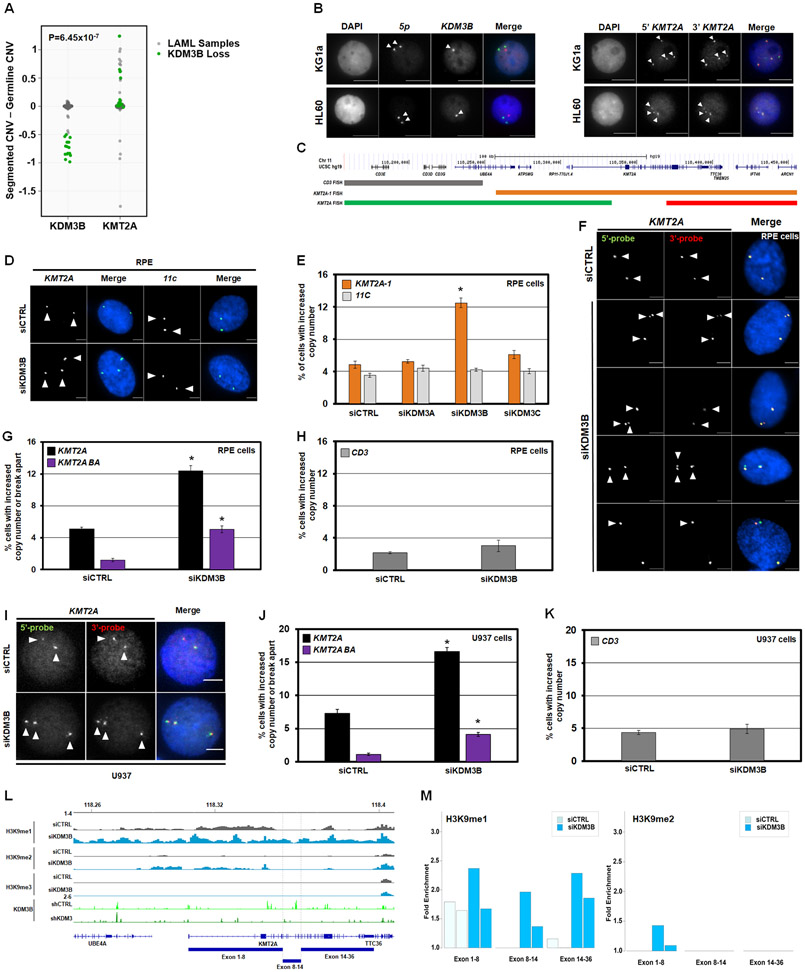
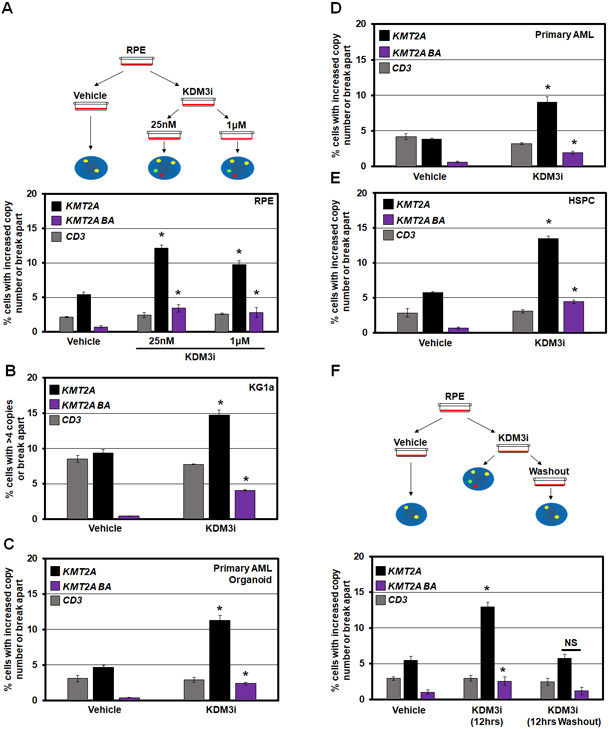
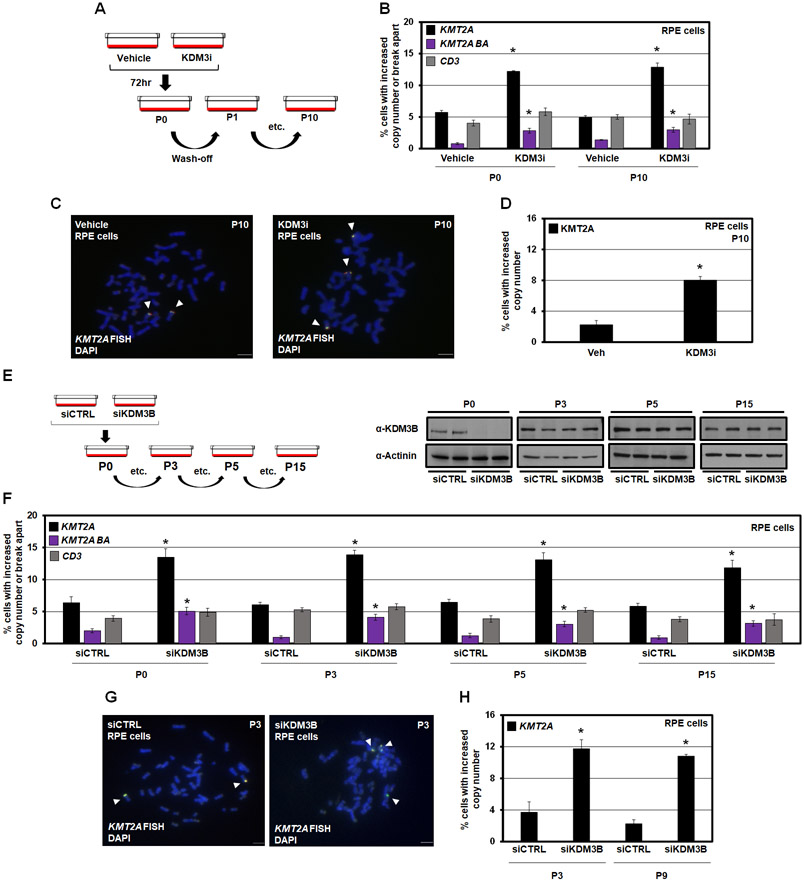
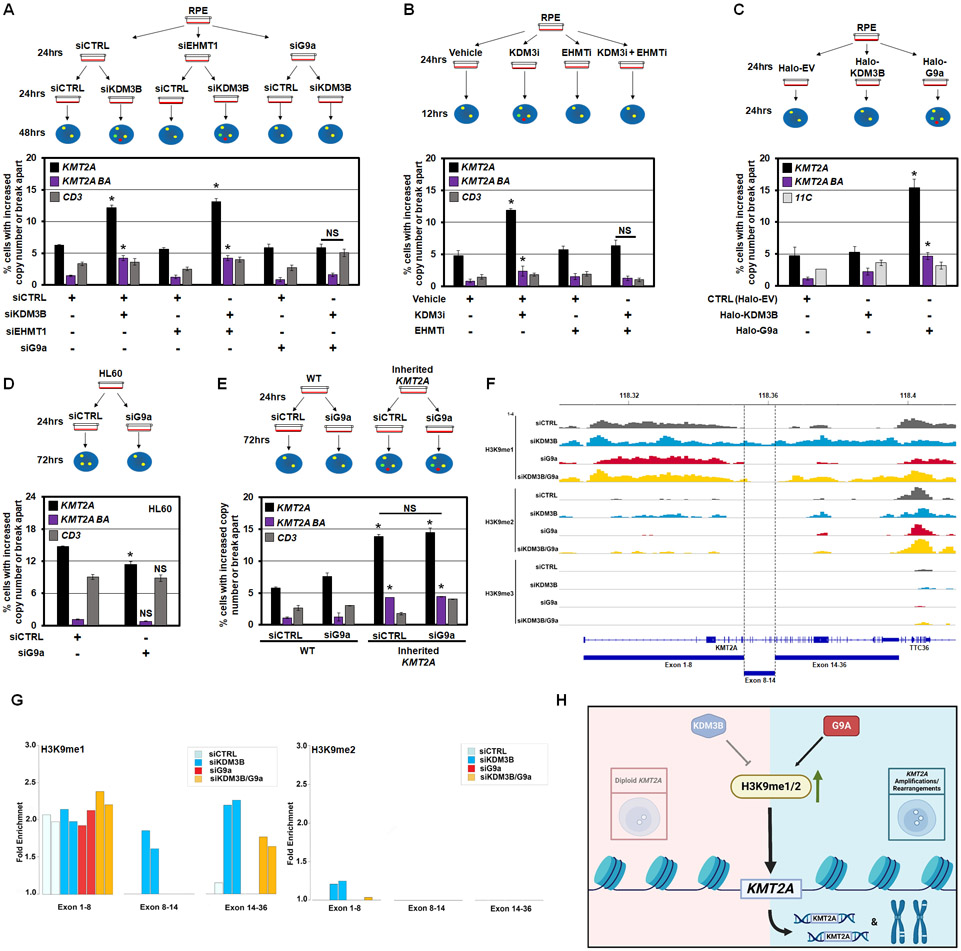
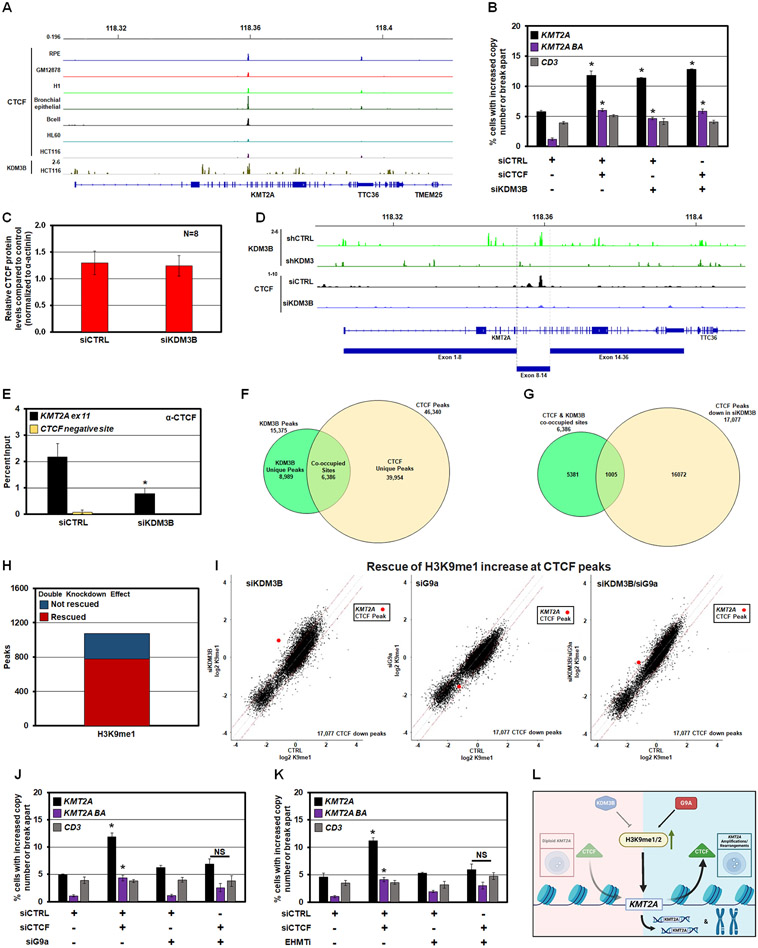
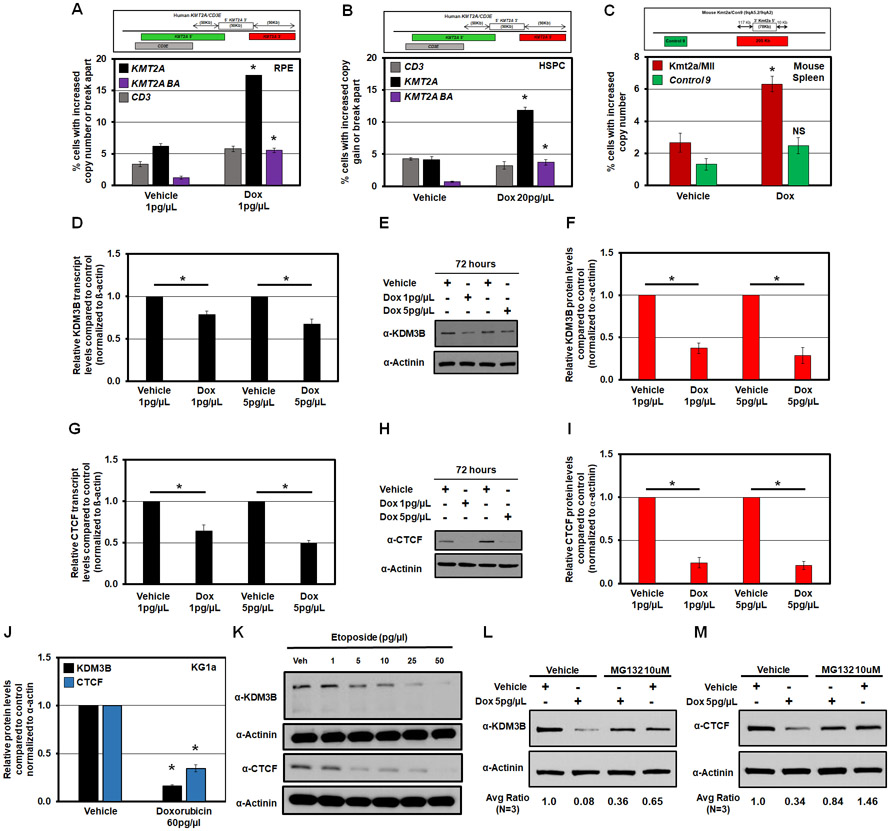
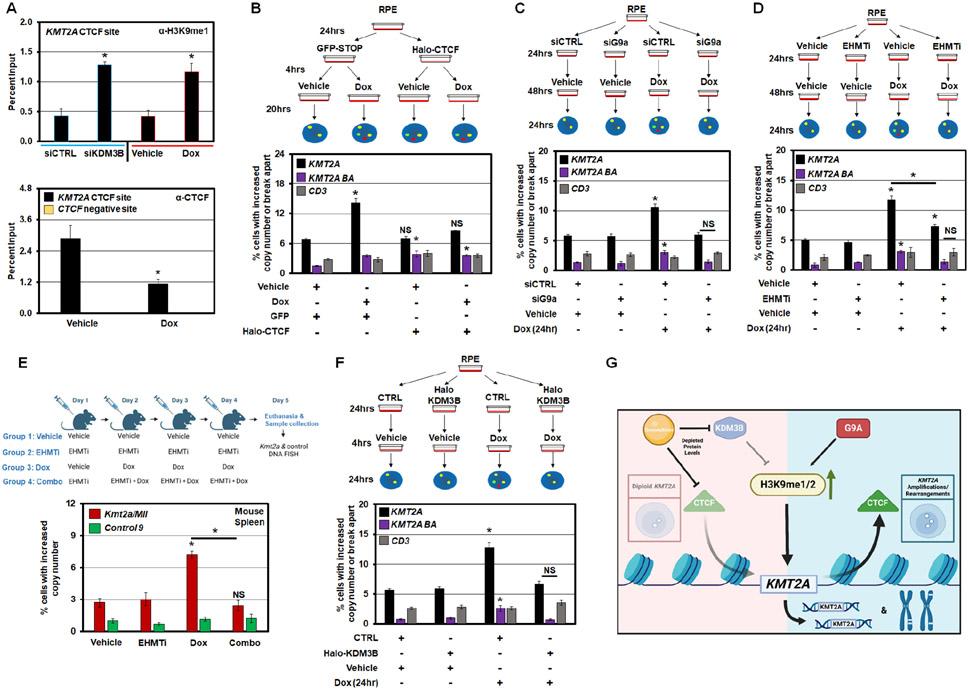
MLL/KMT2A amplifications and translocations are prevalent in infant, adult, and therapy-induced leukemia. However, the molecular contributor(s) to these alterations are unclear. Here, we demonstrate that histone H3 lysine 9 mono- and di-methylation (H3K9me1/2) balance at the MLL/KMT2A locus regulates these amplifications and rearrangements. This balance is controlled by the crosstalk between lysine demethylase KDM3B and methyltransferase G9a/EHMT2. KDM3B depletion increases H3K9me1/2 levels and reduces CTCF occupancy at the MLL/KMT2A locus, in turn promoting amplification and rearrangements. Depleting CTCF is also sufficient to generate these focal alterations. Furthermore, the chemotherapy doxorubicin (Dox), which associates with therapy-induced leukemia and promotes MLL/KMT2A amplifications and rearrangements, suppresses KDM3B and CTCF protein levels. KDM3B and CTCF overexpression rescues Dox-induced MLL/KMT2A alterations. G9a inhibition in human cells or mice also suppresses MLL/KMT2A events accompanying Dox treatment. Therefore, MLL/KMT2A amplifications and rearrangements are controlled by epigenetic regulators that are tractable drug targets, which has clinical implications.
Keywords: CTCF; DNA amplification; G9a; H3K9me; KDM3B; KMT2A; MLL; doxorubicin; ecDNA; rearrangements.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.R.W. has served or is serving as a consultant or advisor for Qsonica, Salarius Pharmaceuticals, Daiichi Sankyo, Inc., Vyne Therapeutics, and Lily Asia Ventures. J.R.W. also receives funding for research from Salarius Pharmaceuticals and Oryzon Genomics. O.G. is a scientific cofounder and shareholder of EpiCypher, Inc., K36 Therapeutics, Inc., and Alternative Bio, Inc. J.J. received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc., and Cullinan Oncology, Inc. J.J. is a cofounder and equity shareholder in Cullgen, Inc., a scientific cofounder and scientific advisory board member of Onsero Therapeutics, Inc., and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc. C.D. receives research funds from Janssen outside the submitted work.
Figures
References
-
- Hanahan D. (2022). Hallmarks of Cancer: New Dimensions. Cancer Discov 12, 31–46. 10.1158/2159-8290.Cd-21-1059. - DOI - PubMed
-
- Song K, Minami JK, Huang A, Dehkordi SR, Lomeli SH, Luebeck J, Goodman MH, Moriceau G, Krijgsman O, Dharanipragada P, et al. (2022). Plasticity of Extrachromosomal and Intrachromosomal BRAF Amplifications in Overcoming Targeted Therapy Dosage Challenges. Cancer Discov 12, 1046–1069. 10.1158/2159-8290.CD-20-0936. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
