

This is a preprint.
Prediction of cancer treatment response from histopathology images through imputed transcriptomics
- PMID: 37790315
- PMCID: PMC10543028
- DOI: 10.21203/rs.3.rs-3193270/v1
Prediction of cancer treatment response from histopathology images through imputed transcriptomics
Update in
-
A deep-learning framework to predict cancer treatment response from histopathology images through imputed transcriptomics.Nat Cancer. 2024 Sep;5(9):1305-1317. doi: 10.1038/s43018-024-00793-2. Epub 2024 Jul 3. Nat Cancer. 2024. PMID: 38961276 Free PMC article.
Abstract
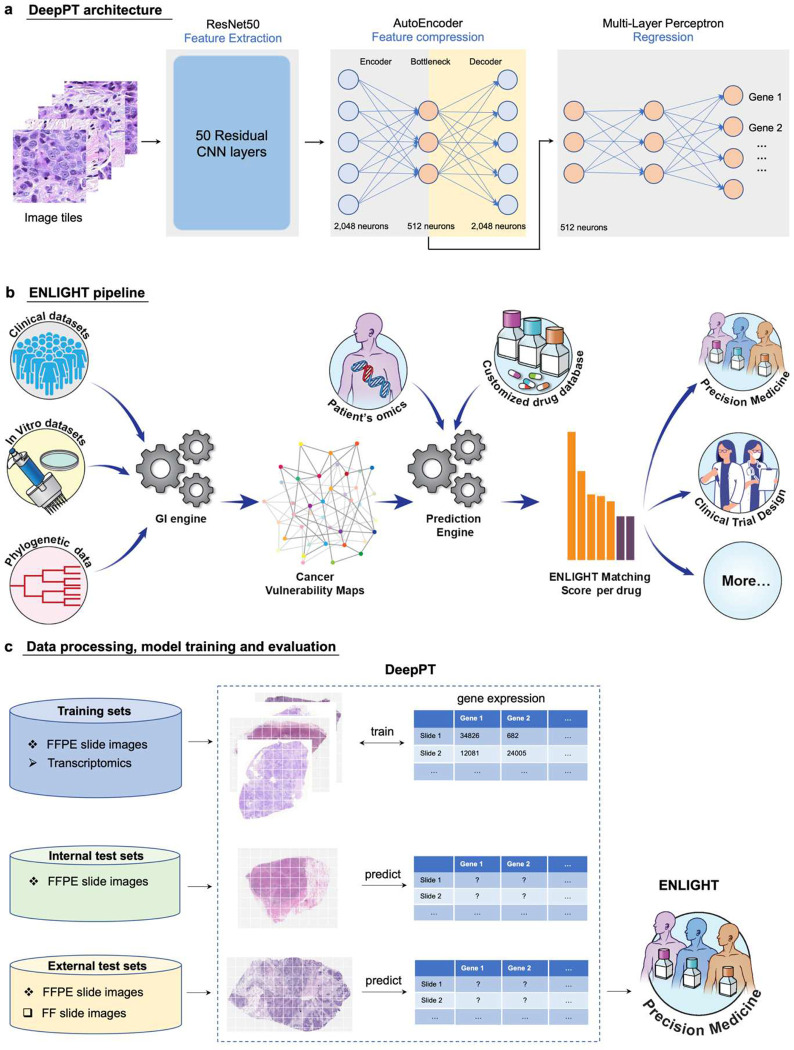
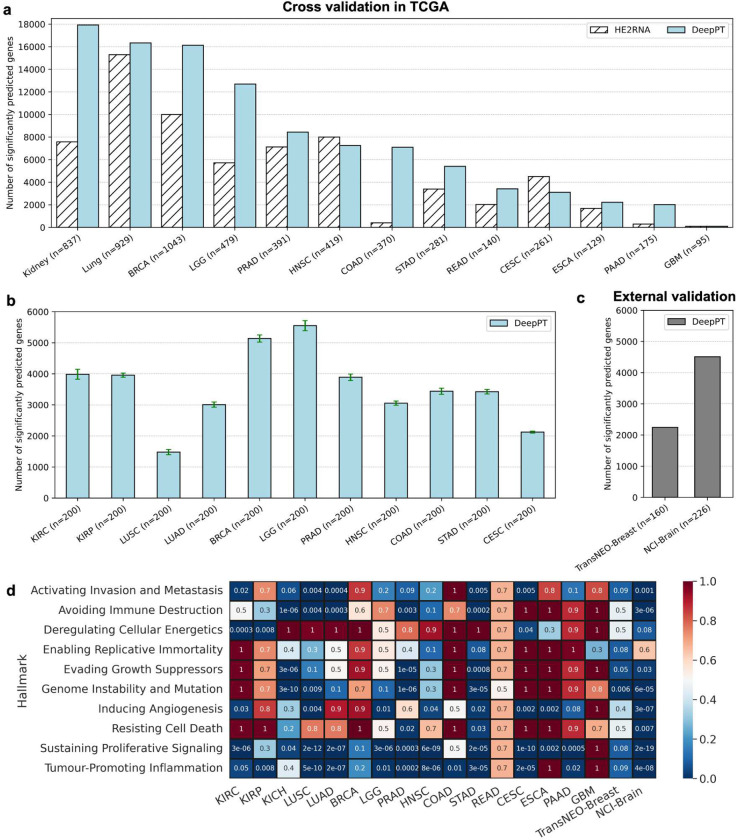
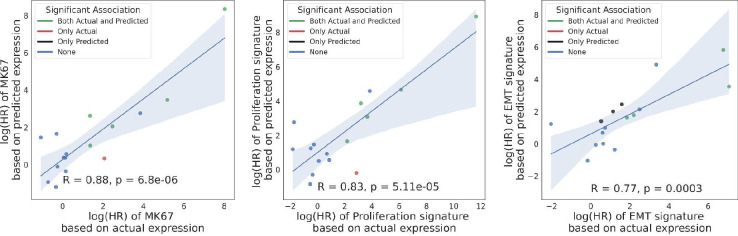
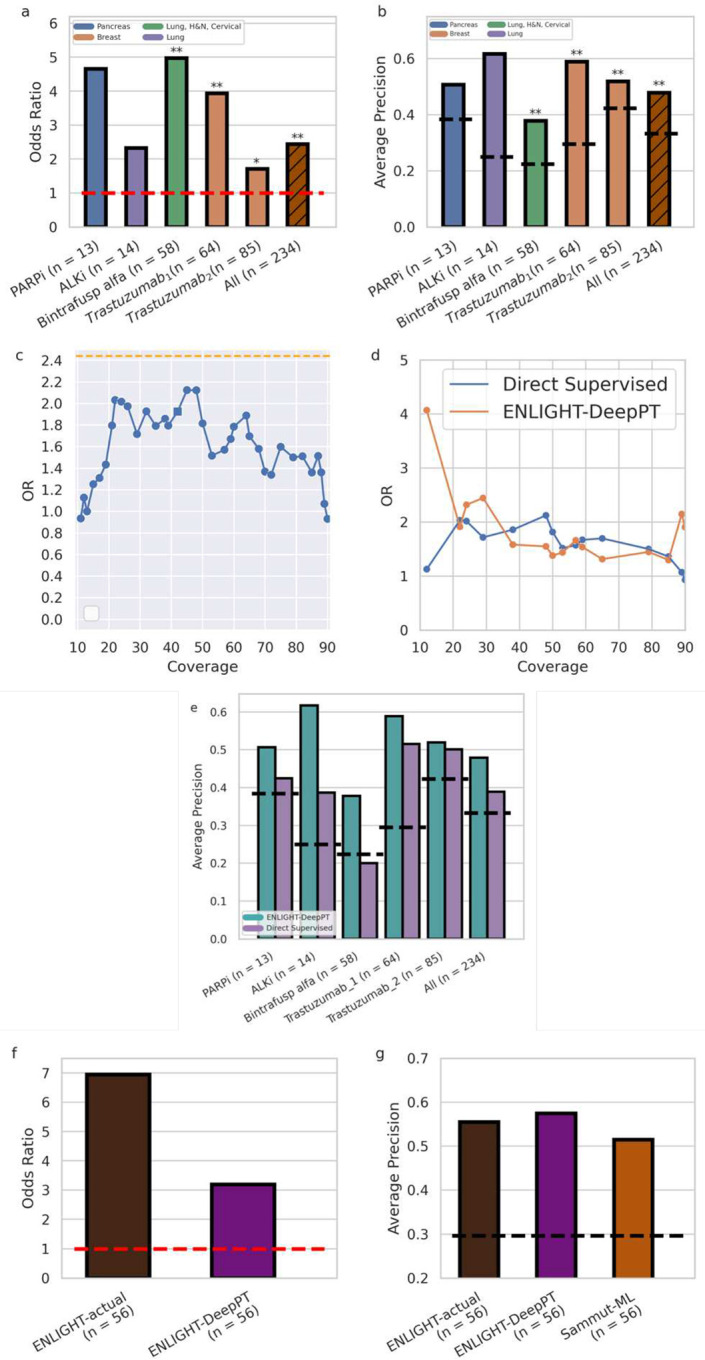
Advances in artificial intelligence have paved the way for leveraging hematoxylin and eosin (H&E)-stained tumor slides for precision oncology. We present ENLIGHT-DeepPT, an approach for predicting response to multiple targeted and immunotherapies from H&E-slides. In difference from existing approaches that aim to predict treatment response directly from the slides, ENLIGHT-DeepPT is an indirect two-step approach consisting of (1) DeepPT, a new deep-learning framework that predicts genome-wide tumor mRNA expression from slides, and (2) ENLIGHT, which predicts response based on the DeepPT inferred expression values. DeepPT successfully predicts transcriptomics in all 16 TCGA cohorts tested and generalizes well to two independent datasets. Our key contribution is showing that ENLIGHT-DeepPT successfully predicts true responders in five independent patients' cohorts involving four different treatments spanning six cancer types with an overall odds ratio of 2.44, increasing the baseline response rate by 43.47% among predicted responders, without the need for any treatment data for training. Furthermore, its prediction accuracy on these datasets is comparable to a supervised approach predicting the response directly from the images, which needs to be trained and tested on the same cohort. ENLIGHT-DeepPT future application could provide clinicians with rapid treatment recommendations to an array of different therapies and importantly, may contribute to advancing precision oncology in developing countries.
Conflict of interest statement
Declaration of interests G.D, D.S.B, E.E, T.B, and R.A are employees of Pangea Biomed. E.R. is a co-founder of Medaware, Metabomed, and Pangea Biomed (divested from the latter). E.R. serves as a non-paid scientific consultant to Pangea Biomed under a collaboration agreement between Pangea Biomed and the NCI.
Figures
References
-
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286: 531–537. - PubMed
-
- Ström P, Kartasalo K, Olsson H, Solorzano L, Delahunt B, Berney DM, et al. Artificial intelligence for diagnosis and grading of prostate cancer in biopsies: a population-based, diagnostic study. Lancet Oncol. 2020;21: 222–232. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
