

This is a preprint.
Role Of The C-C Motif Chemokine Ligand 5 (CCL5) And Its Receptor, C-C Motif Chemokine Receptor 5 (CCR5) In The Genesis Of Aldosterone-induced Hypertension, Vascular Dysfunction, And End-organ Damage
- PMID: 37790434
- PMCID: PMC10542153
- DOI: 10.1101/2023.09.22.558020
Role Of The C-C Motif Chemokine Ligand 5 (CCL5) And Its Receptor, C-C Motif Chemokine Receptor 5 (CCR5) In The Genesis Of Aldosterone-induced Hypertension, Vascular Dysfunction, And End-organ Damage
Update in
-
Role of the CCL5 and Its Receptor, CCR5, in the Genesis of Aldosterone-Induced Hypertension, Vascular Dysfunction, and End-Organ Damage.Hypertension. 2024 Apr;81(4):776-786. doi: 10.1161/HYPERTENSIONAHA.123.21888. Epub 2024 Jan 19. Hypertension. 2024. PMID: 38240165 Free PMC article.
Abstract
Background: Aldosterone, a mineralocorticoid steroid hormone, has been described to initiate cardiovascular diseases by triggering exacerbated sterile vascular inflammation. The functions of C-C Motif Chemokine Ligand 5 (CCL5) and its receptor, C-C Motif Chemokine Receptor 5 (CCR5), are well known in infectious diseases, but their roles in the genesis of aldosterone-induced vascular injury and hypertension are unknown.
Methods: We analyzed the vascular profile, blood pressure, and renal damage in wild-type (CCR5+/+) and CCR5 knockout (CCR5-/-) mice treated with aldosterone (600 μg/kg/day for 14 days) while receiving 1% saline to drink.
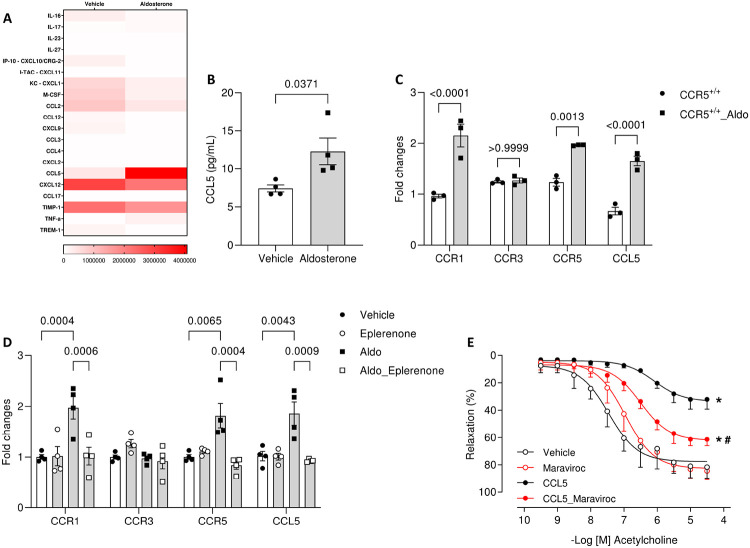
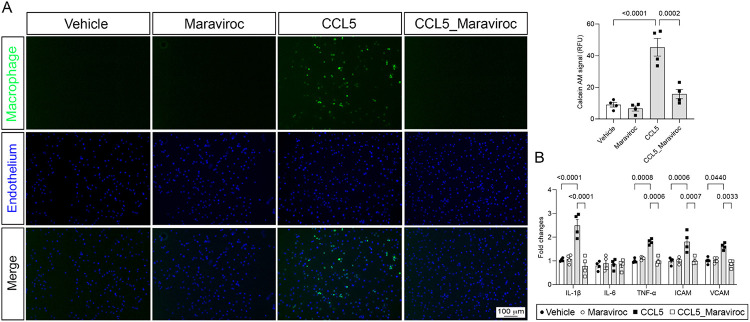
Results: Here, we show that CCR5 plays a central role in aldosterone-induced vascular injury, hypertension, and renal damage. Long-term infusion of aldosterone in CCR5+/+ mice resulted in exaggerated CCL5 circulating levels and vascular CCR5 expression. Aldosterone treatment also triggered vascular injury, characterized by endothelial dysfunction and inflammation, hypertension, and renal damage. Mice lacking CCR5 were protected from aldosterone-induced vascular damage, hypertension, and renal injury. Mechanistically, we demonstrated that CCL5 increased NADPH oxidase 1 (Nox1) expression, reactive oxygen species (ROS) formation, NFκB activation, and inflammation and reduced nitric oxide production in isolated endothelial cells. These effects were abolished by antagonizing CCR5 with Maraviroc. Finally, aortae incubated with CCL5 displayed severe endothelial dysfunction, which is prevented by blocking Nox1, NFκB, or with Maraviroc treatment.
Conclusions: Our data demonstrate that CCL5/CCR5, through activation of NFkB and Nox1, is critically involved in aldosterone-induced vascular and renal damage and hypertension. Our data place CCL5 and CCR5 as potential targets for therapeutic interventions in conditions with aldosterone excess.
Keywords: NADPH oxidases; aldosterone; chemokines; chemokines receptors; oxidative stress.
Figures




References
-
- Scott JH, Menouar MA, Dunn RJ. Physiology, aldosterone. Statpearls. Treasure Island (FL); 2023. - PubMed
-
- Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y, Correa JW, Gagnon AM, Gomez-Sanchez CE, Gomez-Sanchez EP, Sorisky A, Ooi TC, Ruzicka M, Burns KD, Touyz RM. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: Implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension. 2012;59:1069–1078 - PubMed
-
- Dinh Cat AN, Friederich-Persson M, White A, Touyz RM. Adipocytes, aldosterone and obesity-related hypertension. J Mol Endocrinol. 2016;57:F7–F21 - PubMed
-
- Calhoun DA. Aldosterone and cardiovascular disease: Smoke and fire. Circulation. 2006;114:2572–2574 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources