

This is a preprint.
Evaluation of large language models for discovery of gene set function
- PMID: 37790547
- PMCID: PMC10543283
- DOI: 10.21203/rs.3.rs-3270331/v1
Evaluation of large language models for discovery of gene set function
Update in
-
Evaluation of large language models for discovery of gene set function.Nat Methods. 2025 Jan;22(1):82-91. doi: 10.1038/s41592-024-02525-x. Epub 2024 Nov 28. Nat Methods. 2025. PMID: 39609565 Free PMC article.
Abstract
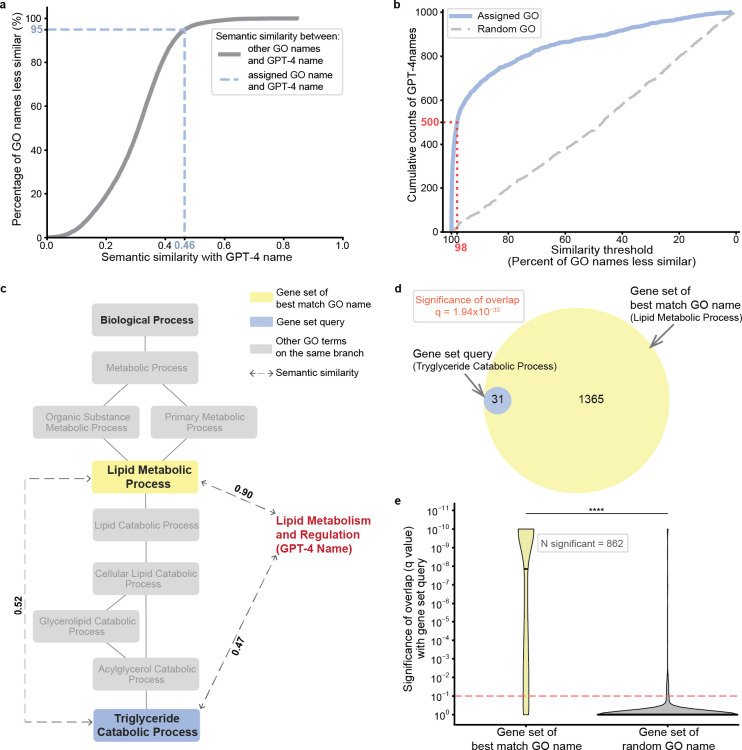
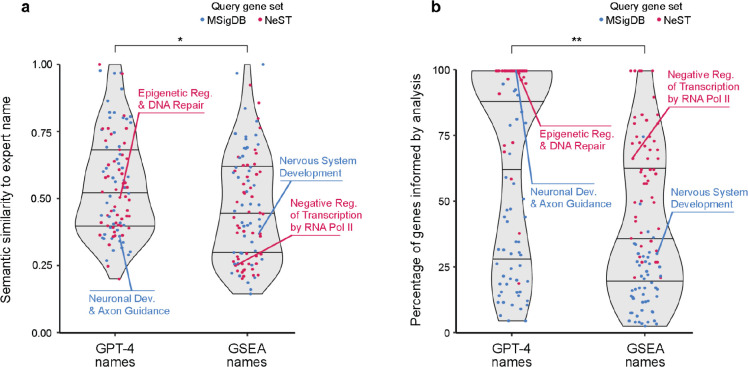
Gene set analysis is a mainstay of functional genomics, but it relies on manually curated databases of gene functions that are incomplete and unaware of biological context. Here we evaluate the ability of OpenAI's GPT-4, a Large Language Model (LLM), to develop hypotheses about common gene functions from its embedded biomedical knowledge. We created a GPT-4 pipeline to label gene sets with names that summarize their consensus functions, substantiated by analysis text and citations. Benchmarking against named gene sets in the Gene Ontology, GPT-4 generated very similar names in 50% of cases, while in most remaining cases it recovered the name of a more general concept. In gene sets discovered in 'omics data, GPT-4 names were more informative than gene set enrichment, with supporting statements and citations that largely verified in human review. The ability to rapidly synthesize common gene functions positions LLMs as valuable functional genomics assistants.
Figures



References
-
- Huang D. W., Sherman B. T. & Lempicki R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
