

JNKs protect from cholestatic liver disease progression by modulating Apelin signalling
- PMID: 37791376
- PMCID: PMC10543210
- DOI: 10.1016/j.jhepr.2023.100854
JNKs protect from cholestatic liver disease progression by modulating Apelin signalling
Abstract
Background & aims: Cholestatic liver injury is associated with c-Jun N-terminal kinases (JNK) activation in distinct cell types. Its hepatocyte-specific function during cholestasis, however, has not yet been established. Therefore, in our present study, we investigated the role of JNK1/2 during cholestasis and dissected its hepatocyte-specific function.
Methods: A cohort of patients with primary biliary cholangitis (n = 29) and primary sclerosing cholangitis (n = 37) was examined. Wild-type, hepatocyte-specific knockout mice for Jnk2 (Jnk2Δhepa) or Jnk1 and Jnk2 (Jnk1Δhepa/2Δhepa) were generated. Mice were subjected to bile duct ligation (BDL) or carbon tetrachloride (CCl4) treatment. Finally, Apelin signalling was blocked using a specific inhibitor. As an interventional approach, Jnk1/2 were silenced in wild-type mice using lipid nanoparticles for small interfering RNA delivery.
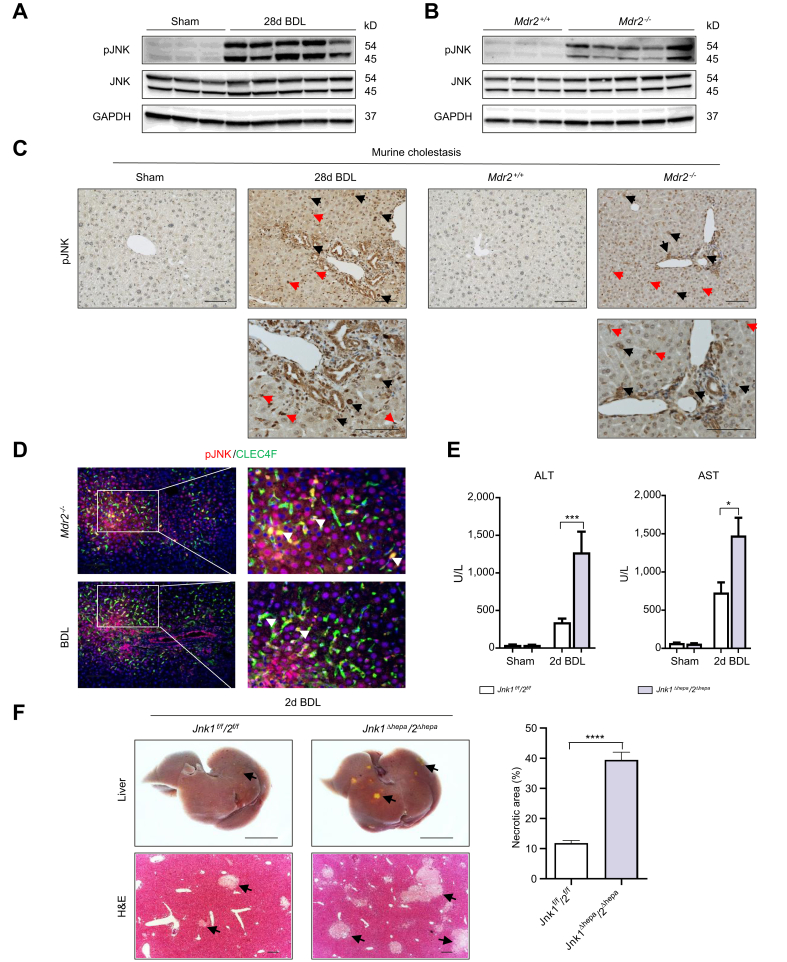
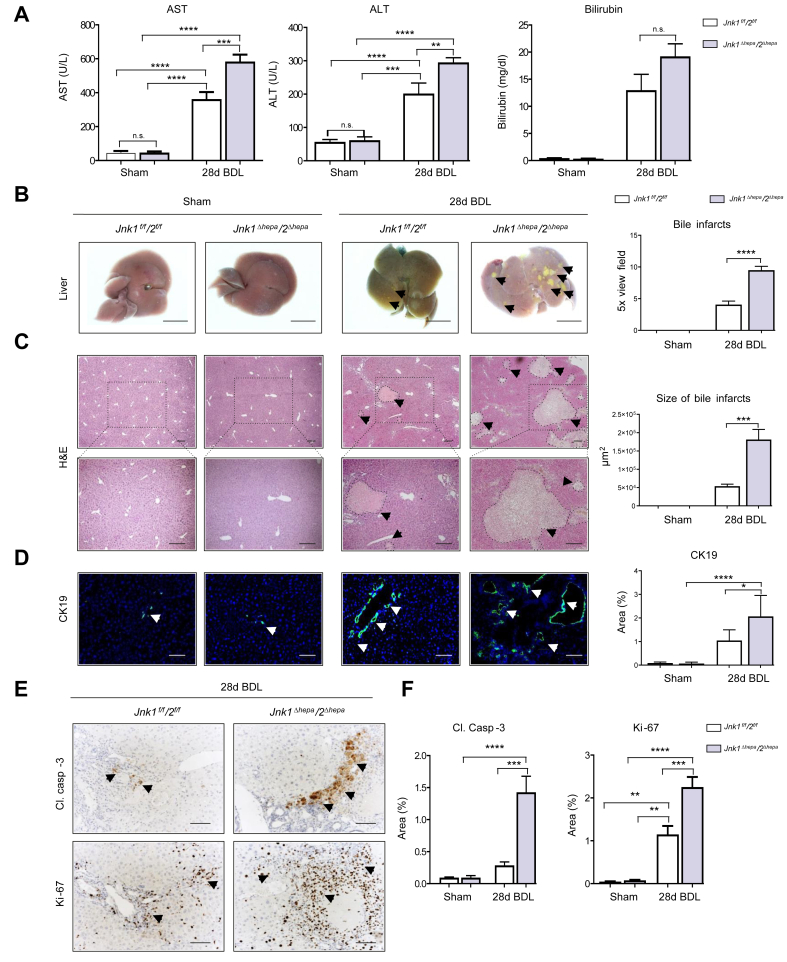
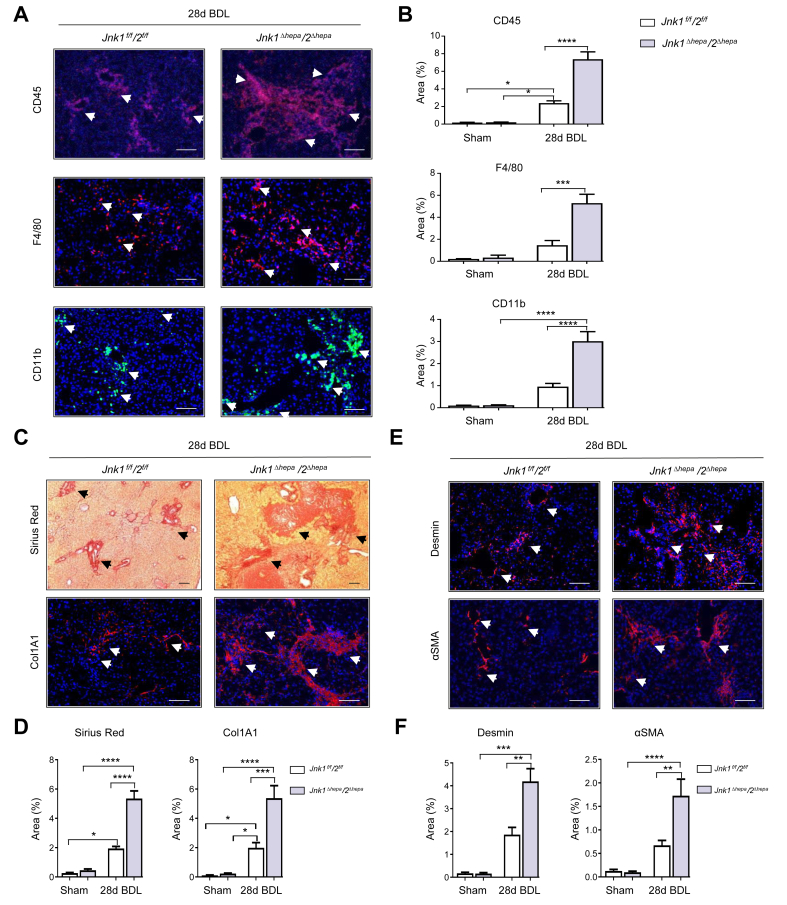
Results: JNK activation was increased in liver specimens from patients with chronic cholestasis (primary biliary cholangitis and primary sclerosing cholangitis) and in livers of Mdr2-/- and BDL-treated animals. In Jnk1Δhepa/2Δhepa animals, serum transaminases increased after BDL, and liver histology demonstrated enhanced cell death, compensatory proliferation, hepatic fibrogenesis, and inflammation. Furthermore, microarray analysis revealed that hepatocytic Jnk1/2 ablation induces JNK-target genes involved in oxidative stress and Apelin signalling after BDL. Consequently, blocking Apelin signalling attenuated BDL-induced liver injury and fibrosis in Jnk1Δhepa/2Δhepa mice. Finally, we established an interventional small interfering RNA approach of selective Jnk1/2 targeting in hepatocytes in vivo, further demonstrating the essential protective role of Jnk1/2 during cholestasis.
Conclusions: Jnk1 and Jnk2 work together to protect hepatocytes from cholestatic liver disease by controlling Apelin signalling. Dual modification of JNK signalling in hepatocytes is feasible, and enhancing its expression might be an attractive therapeutic approach for cholestatic liver disease.
Impact and implications: The cell-specific function of Jnk genes during cholestasis has not been explicitly explored. In this study, we showed that combined Jnk1/2, but not Jnk2 deficiency, in hepatocytes exacerbates liver damage and fibrosis by enhancing Apelin signalling, which contributes to cholestasis progression. Combined cell-specific Jnk targeting may be a new molecular strategy for treating cholestatic liver disease.
Keywords: Apelin; Cholestasis; Fibrosis; Hepatocytes; c-Jun N-terminal kinases (JNK).
© 2023 The Authors.
Conflict of interest statement
The authors declare that they have no financial competing interests. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures





References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
