

Rare variant associations with plasma protein levels in the UK Biobank
- PMID: 37794183
- PMCID: PMC10567546
- DOI: 10.1038/s41586-023-06547-x
Rare variant associations with plasma protein levels in the UK Biobank
Abstract
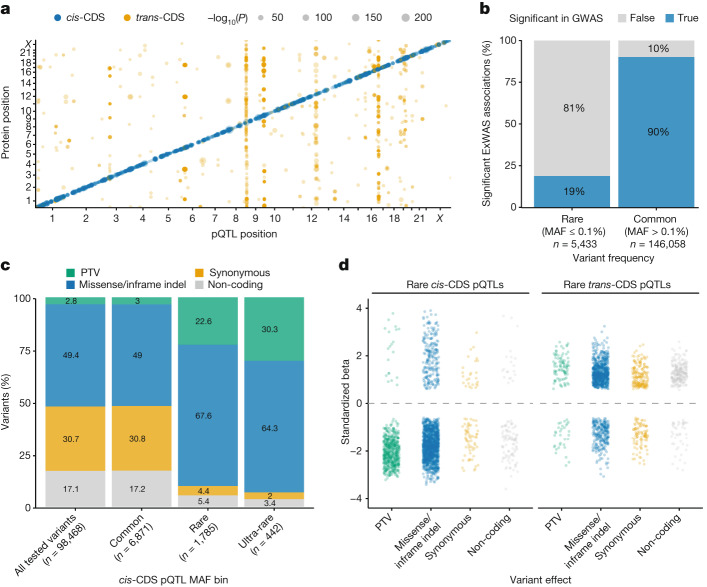
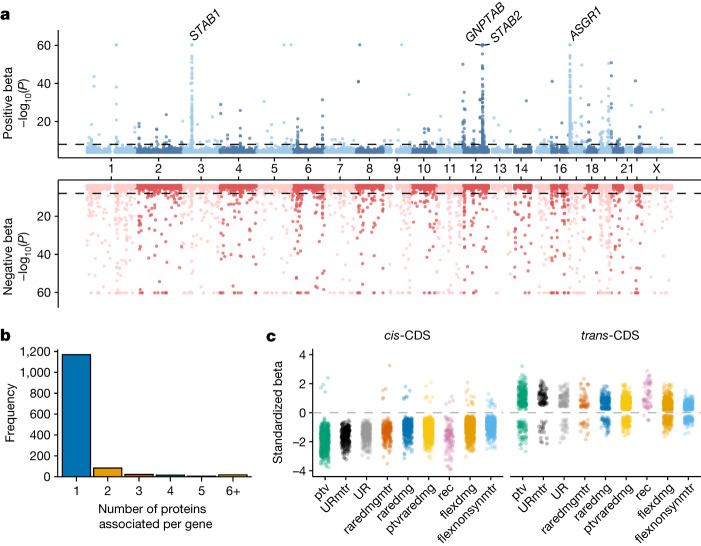
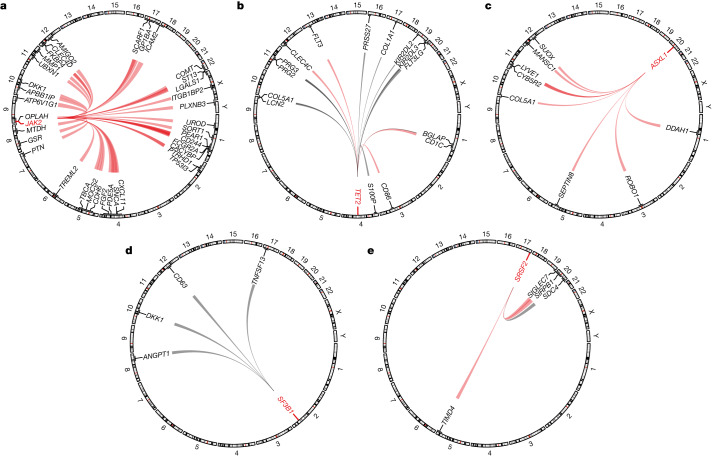
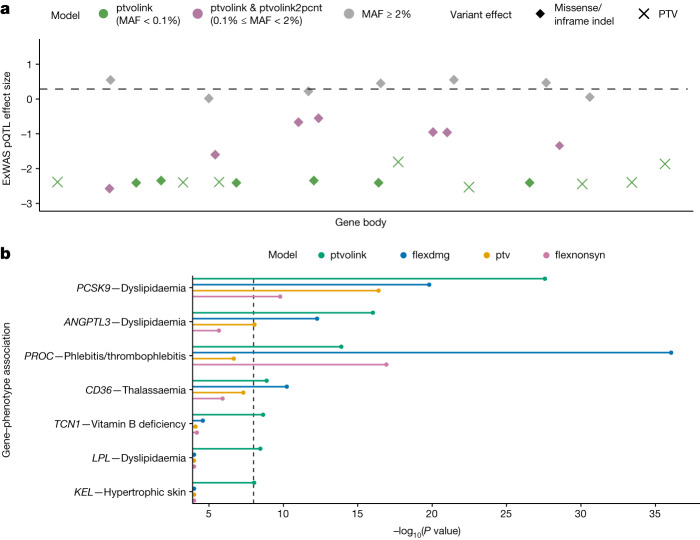
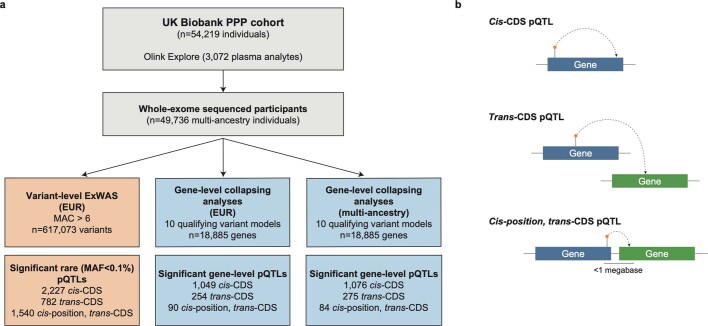
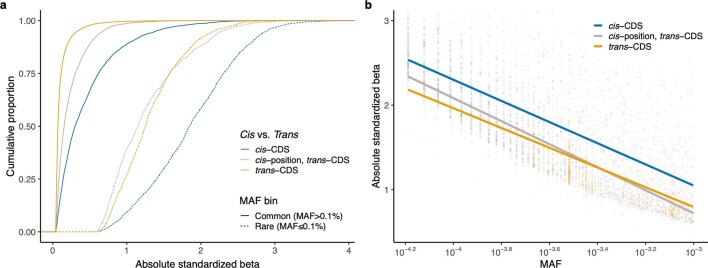
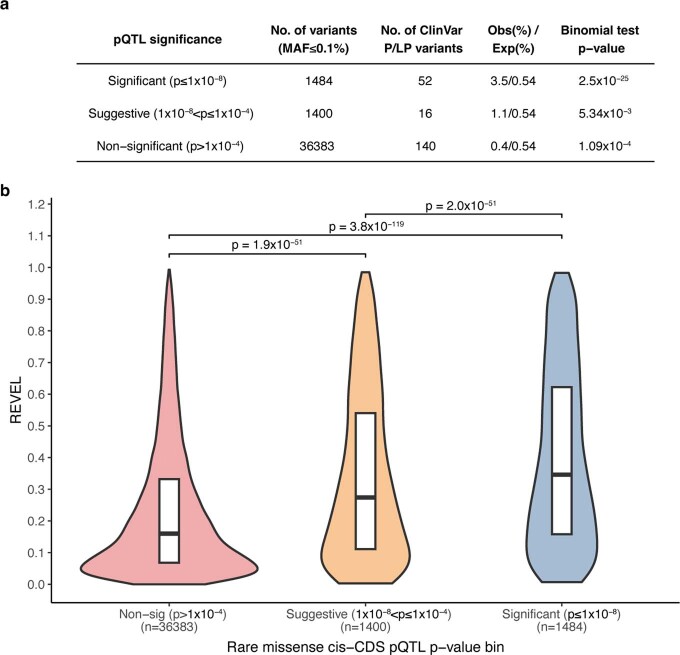
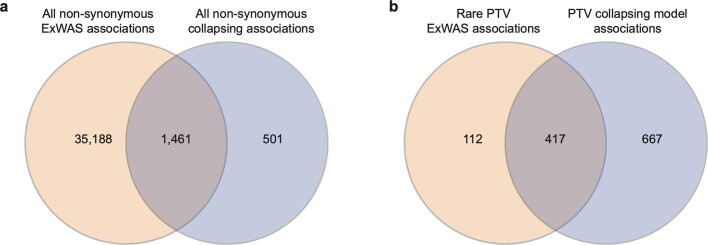
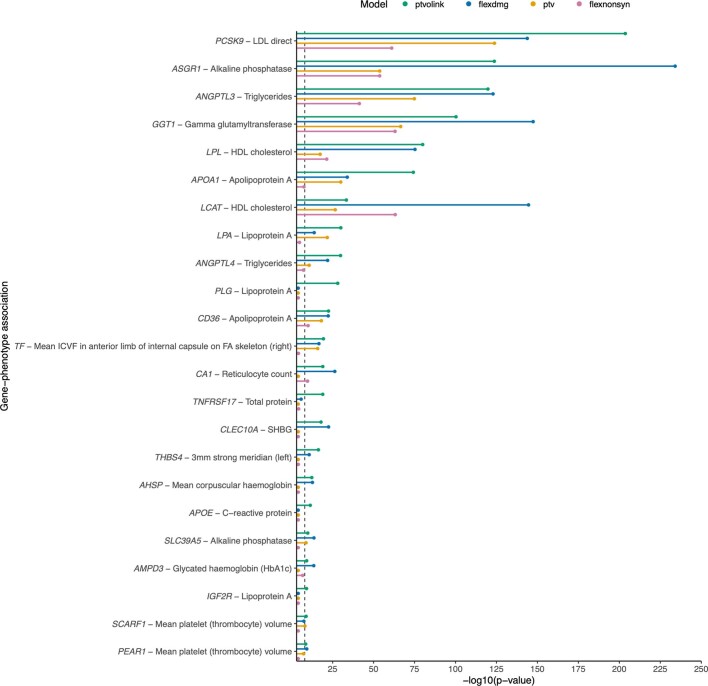
Integrating human genomics and proteomics can help elucidate disease mechanisms, identify clinical biomarkers and discover drug targets1-4. Because previous proteogenomic studies have focused on common variation via genome-wide association studies, the contribution of rare variants to the plasma proteome remains largely unknown. Here we identify associations between rare protein-coding variants and 2,923 plasma protein abundances measured in 49,736 UK Biobank individuals. Our variant-level exome-wide association study identified 5,433 rare genotype-protein associations, of which 81% were undetected in a previous genome-wide association study of the same cohort5. We then looked at aggregate signals using gene-level collapsing analysis, which revealed 1,962 gene-protein associations. Of the 691 gene-level signals from protein-truncating variants, 99.4% were associated with decreased protein levels. STAB1 and STAB2, encoding scavenger receptors involved in plasma protein clearance, emerged as pleiotropic loci, with 77 and 41 protein associations, respectively. We demonstrate the utility of our publicly accessible resource through several applications. These include detailing an allelic series in NLRC4, identifying potential biomarkers for a fatty liver disease-associated variant in HSD17B13 and bolstering phenome-wide association studies by integrating protein quantitative trait loci with protein-truncating variants in collapsing analyses. Finally, we uncover distinct proteomic consequences of clonal haematopoiesis (CH), including an association between TET2-CH and increased FLT3 levels. Our results highlight a considerable role for rare variation in plasma protein abundance and the value of proteogenomics in therapeutic discovery.
© 2023. The Author(s).
Conflict of interest statement
R.S.D., O.S.B., B.P.P., D. Matelska, E.W., J.M., E.O., V.A.H., K.R.S., K.C., S.W., A.R.H., D.S.P., M.A.F., C.V., B.C., A.P., D.V., M.N.P., Q.W. and S.P. are current employees and/or stockholders of AstraZeneca. B.B.S., C.D.W. and H.R. are employees and/or stockholders of Biogen. E.A.A. is a founder of Personalis, Inc., DeepCell, Inc. and Svexa Inc.; a founding advisor of Nuevocor; a non-executive director at AstraZeneca; and an advisor to SequenceBio, Novartis, Medical Excellence Capital, Foresite Capital and Third Rock Ventures.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
