

Reconciling intrinsic properties of activating TNF receptors by native ligands versus synthetic agonists
- PMID: 37795079
- PMCID: PMC10546206
- DOI: 10.3389/fimmu.2023.1236332
Reconciling intrinsic properties of activating TNF receptors by native ligands versus synthetic agonists
Abstract
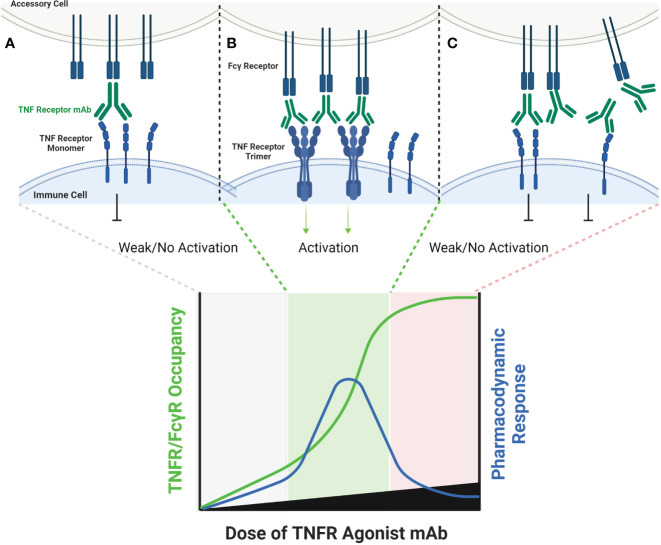
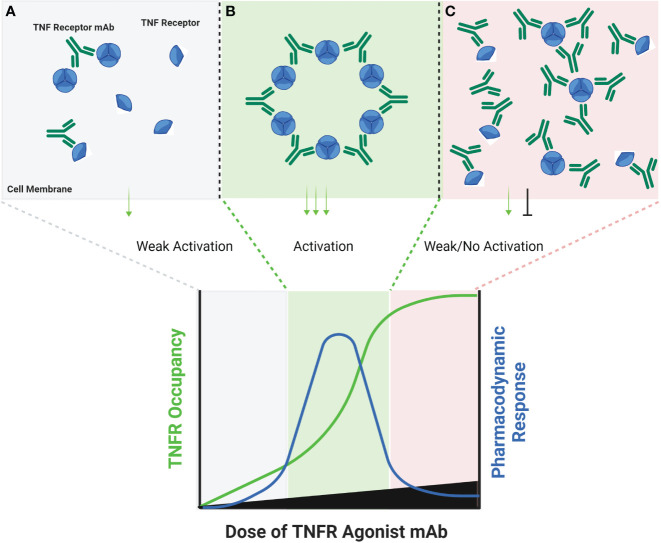
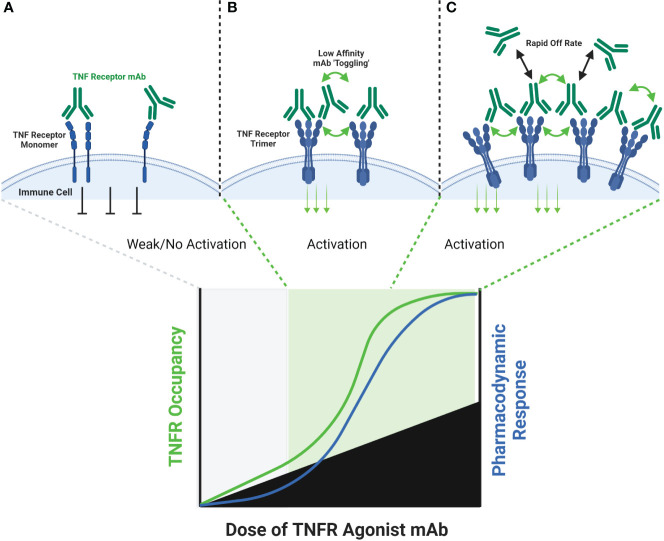
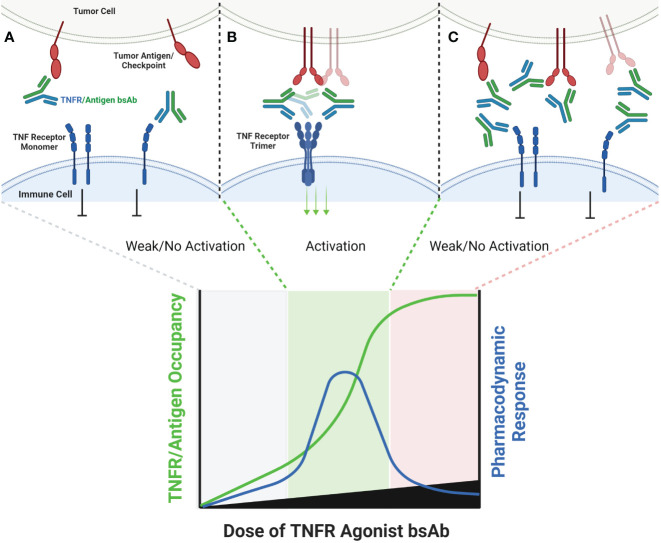
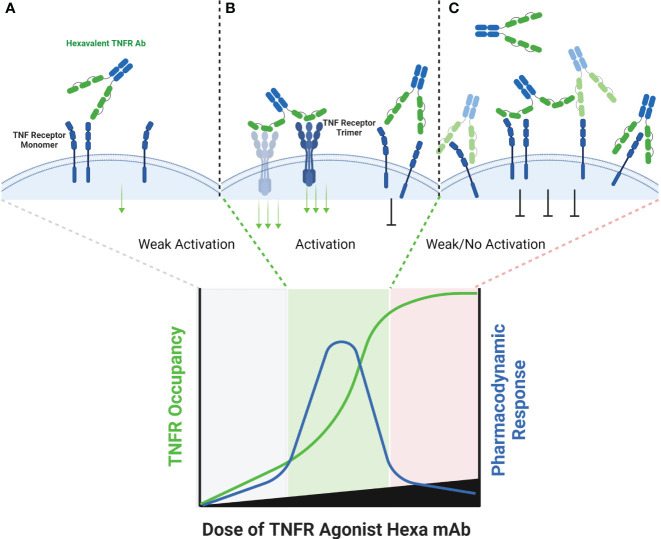
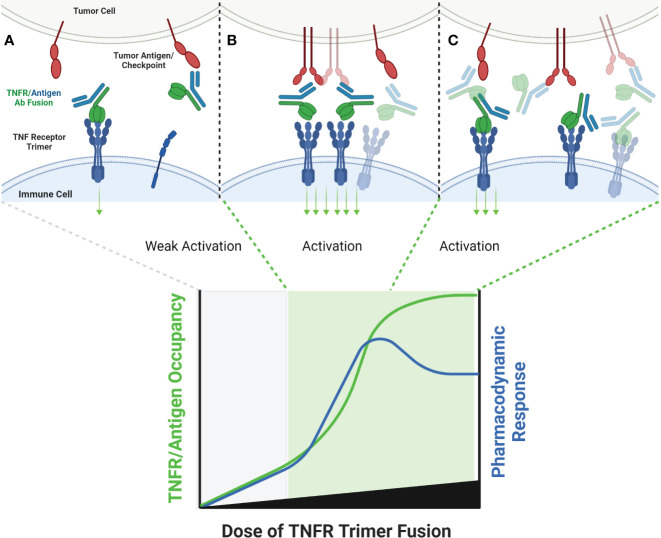
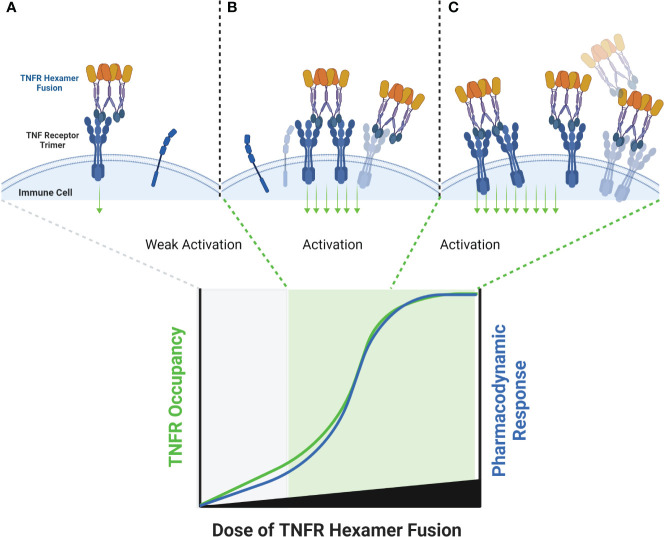
The extracellular domain of tumor necrosis factor receptors (TNFR) generally require assembly into a homotrimeric quaternary structure as a prerequisite for initiation of signaling via the cytoplasmic domains. TNF receptor homotrimers are natively activated by similarly homo-trimerized TNF ligands, but can also be activated by synthetic agonists including engineered antibodies and Fc-ligand fusion proteins. A large body of literature from pre-clinical models supports the hypothesis that synthetic agonists targeting a diverse range of TNF receptors (including 4-1BB, CD40, OX40, GITR, DR5, TNFRSF25, HVEM, LTβR, CD27, and CD30) could amplify immune responses to provide clinical benefit in patients with infectious diseases or cancer. Unfortunately, however, the pre-clinical attributes of synthetic TNF receptor agonists have not translated well in human clinical studies, and have instead raised fundamental questions regarding the intrinsic biology of TNF receptors. Clinical observations of bell-shaped dose response curves have led some to hypothesize that TNF receptor overstimulation is possible and can lead to anergy and/or activation induced cell death of target cells. Safety issues including liver toxicity and cytokine release syndrome have also been observed in humans, raising questions as to whether those toxicities are driven by overstimulation of the targeted TNF receptor, a non-TNF receptor related attribute of the synthetic agonist, or both. Together, these clinical findings have limited the development of many TNF receptor agonists, and may have prevented generation of clinical data which reflects the full potential of TNF receptor agonism. A number of recent studies have provided structural insights into how different TNF receptor agonists bind and cluster TNF receptors, and these insights aid in deconvoluting the intrinsic biology of TNF receptors with the mechanistic underpinnings of synthetic TNF receptor agonist therapeutics.
Keywords: 41BB; CD40; OX40; TNF ligand; TNF receptor; TNF superfamily; agonist.
Copyright © 2023 Fromm, de Silva and Schreiber.
Conflict of interest statement
All authors are shareholders and employed by the company Shattuck Labs, Inc.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials