

Hemoglobin profile and molecular characteristics of the complex interaction of hemoglobin Doi-Saket [α9(A7) asn > lys, HBA2:c.30C > a], a novel α2α1 hybrid globin variant, with hemoglobin E [β26(B8) Glu > lys, HBB:c.79G > A] and deletional α+-thalassemia in a Thai family
- PMID: 37796611
- PMCID: PMC10557546
- DOI: 10.1080/07853890.2023.2264174
Hemoglobin profile and molecular characteristics of the complex interaction of hemoglobin Doi-Saket [α9(A7) asn > lys, HBA2:c.30C > a], a novel α2α1 hybrid globin variant, with hemoglobin E [β26(B8) Glu > lys, HBB:c.79G > A] and deletional α+-thalassemia in a Thai family
Abstract
Background: An increasing number of α-hemoglobin (Hb) variants is causing various clinical symptoms; therefore, accurate identification of these Hb variants is important.
Objective: This study aimed to describe the molecular and hematological characteristics of novel Hb Doi-Saket that gives rise to a typical α+-thalassemia phenotype in carriers with and without other hemoglobinopathies.
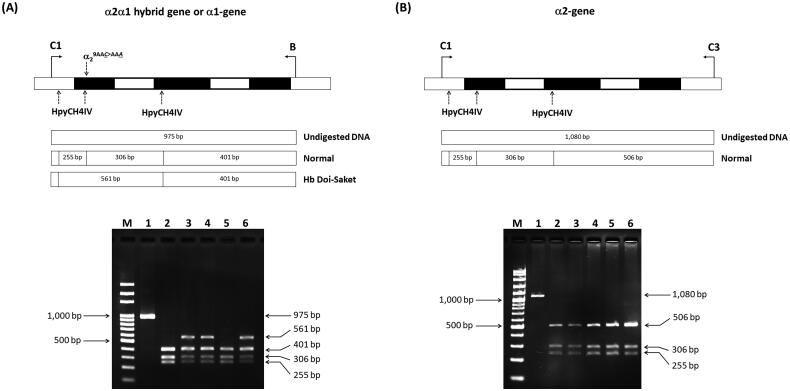
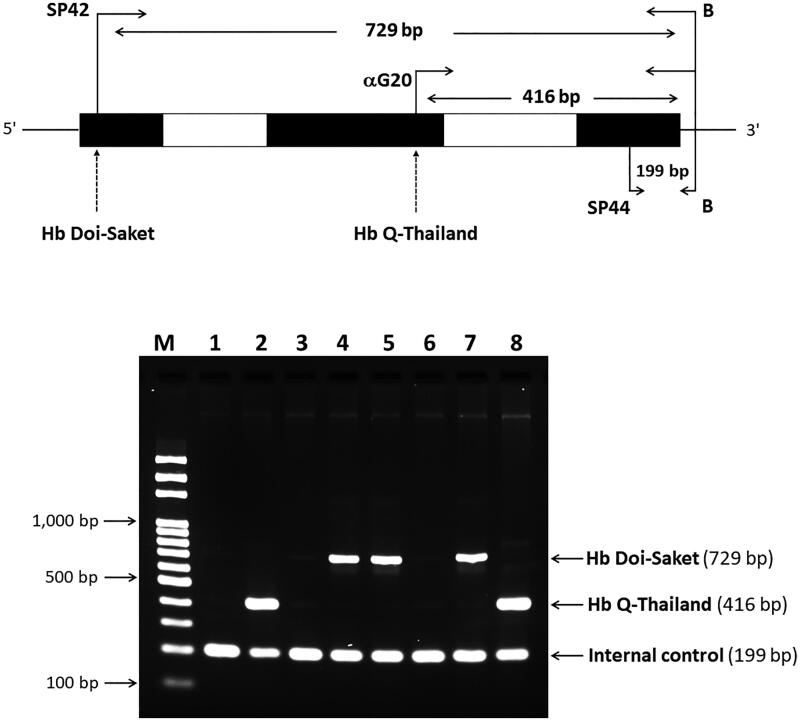
Materials and methods: Biological samples from a proband and his family members were analyzed. Hematological profiles were analyzed using a standard automated cell counter. Hb was analyzed by capillary electrophoresis and high-performance liquid chromatography. Mutations and globin haplotype were identified by DNA analysis. Novel diagnostic tools based on allele-specific polymerase chain reaction (PCR) and PCR-restriction fragment length polymorphism were developed.
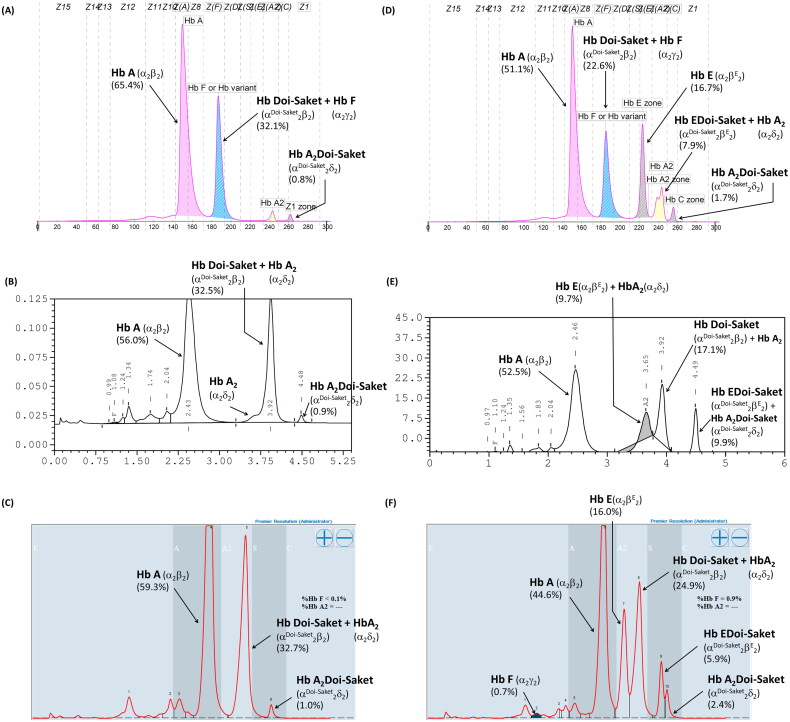
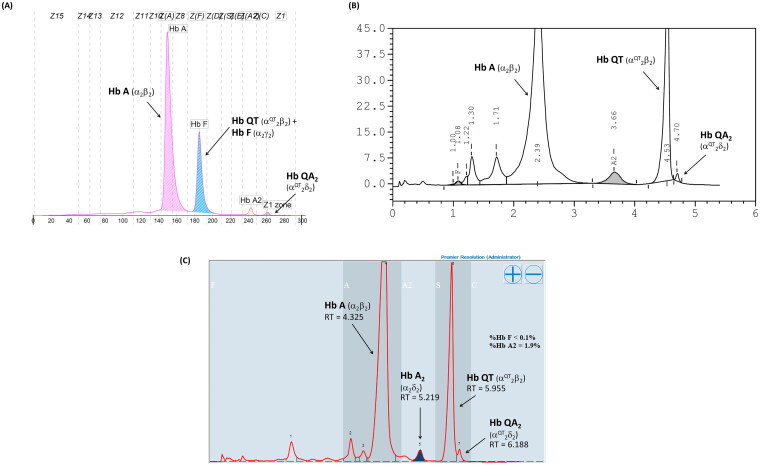
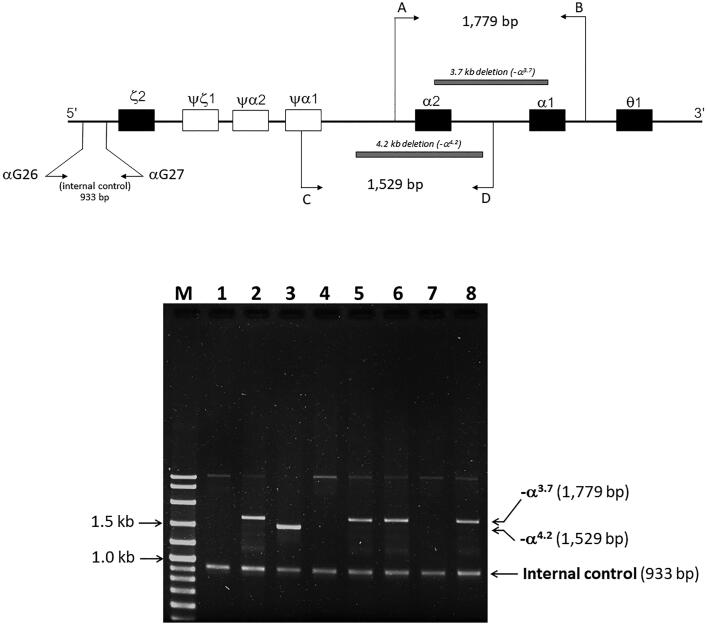
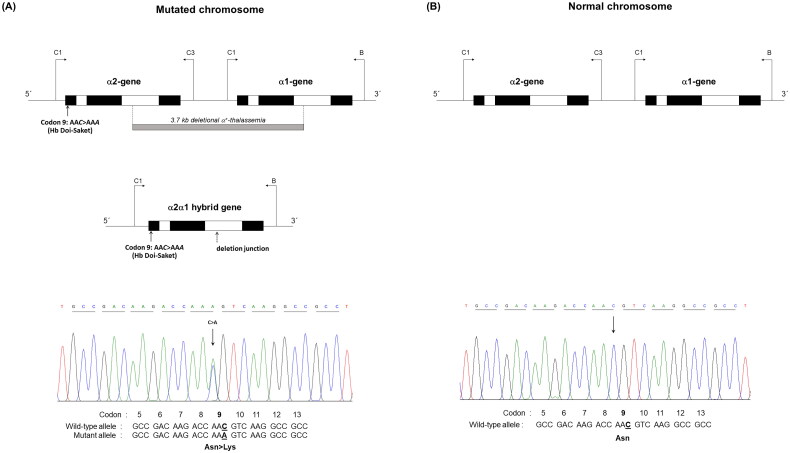
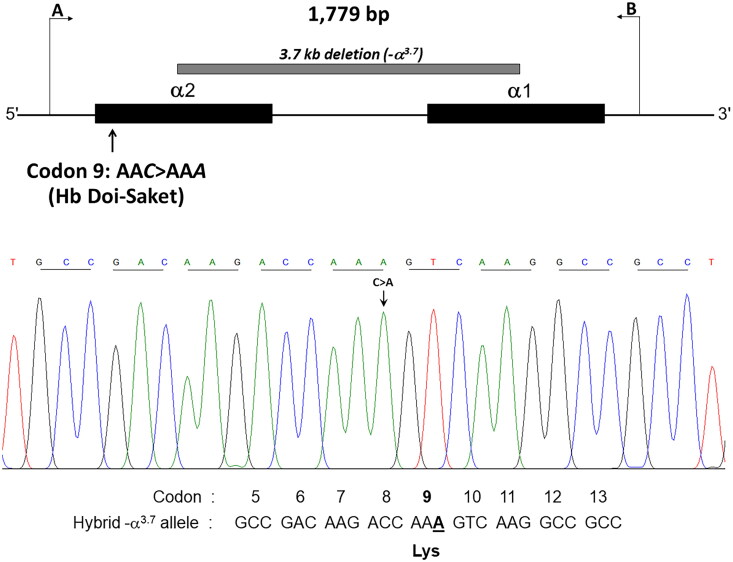
Results: Hb analysis showed a major abnormal Hb fraction, moving slower than HbA, and a minor Hb fraction alongside HbA2 in the proband, his father, and son. DNA analysis of the α-globin gene identified the -α3.7 deletion and in cis the C > A mutation on codon 9 of the α2α1 gene, corresponding to Hb Doi-Saket [α9(A7) Asn > Lys]. This mutation could be identified using newly developed allele-specific PCR-based assays. The Hb Doi-Saket al.lele was significantly associated with haplotype [- + M + + 0 -]. Interaction of αDoi-Saket with βE globin chains led to a new Hb variant (HbE Doi-Saket). Phenotypic expression was clinically silent in heterozygotes and might present slight microcytosis.
Conclusions: Hb Doi-Saket emphasizes a great diversity present in α-globin gene. The mutation in this family from Thailand was linked to -α3.7 and caused mild microcytosis in the carriers. The combination of this variant with deletions in α genes might cause a severe clinical phenotype. Different methods of separation can provide useful information in diagnosis, and a complete molecular approach is needed for confirmation before considering patient management.
Keywords: Hb Doi-Saket; HbE; Thailand; α+-thalassemia; α-globin variant.
Plain language summary
The Hb Doi-Saket is a novel α-globin variant mutation occurring in the α2-globin gene in cis to the -α3.7 kb chromosome.The carrier of Hb Doi-Saket may present slight microcytosis and have severe clinical entities when it interacts with deletions in α-globin genes.Hb analysis with the HPLC system could completely separate Hb Doi-Saket and its derivative from other Hbs.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
Similar articles
-
Ten Years of Routine α- and β-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations.Hemoglobin. 2016;40(2):75-84. doi: 10.3109/03630269.2015.1113990. Epub 2015 Dec 4. Hemoglobin. 2016. PMID: 26635043 Review.
-
Phenotypic Expression of Known and Novel Hemoglobin A2-Variants, Hemoglobin A2-Mae Phrik [Delta 52(D3) Asp > Gly, HBD:c.158A > G], Associated with Hemoglobin E [Beta 26(B8) Glu > Lys, HBB:c.79G > A] in Thailand.Genes (Basel). 2022 May 27;13(6):959. doi: 10.3390/genes13060959. Genes (Basel). 2022. PMID: 35741722 Free PMC article.
-
Complex interaction of Hb Hekinan [alpha27(B8) Glu-Asp] and Hb E [beta26(B8) Glu-Lys] with a deletional alpha-thalassemia 1 in a Thai family.Eur J Haematol. 2003 May;70(5):304-9. doi: 10.1034/j.1600-0609.2003.00049.x. Eur J Haematol. 2003. PMID: 12694166
-
Molecular Characterization and Hematological Aspects of Hb E-Myanmar [β26(B8)Glu→Lys and β65(E9)Lys→Asn, HBB: c.[79G>A;198G>C]): A Novel β-Thalassemic Hemoglobin Variant.Hemoglobin. 2020 Nov;44(6):385-390. doi: 10.1080/03630269.2020.1848860. Epub 2020 Nov 22. Hemoglobin. 2020. PMID: 33222574
-
Characterization and Confirmation of Mildly Unstable Hb Pontoise or α1 63(E12) Ala > Asp and Literature Review.Hemoglobin. 2025 Jan;49(1):26-30. doi: 10.1080/03630269.2025.2451411. Epub 2025 Jan 20. Hemoglobin. 2025. PMID: 39833127 Review.
References
-
- Svasti J, Srisomsap C, Winichagoon P, et al. . Detection and structural analysis of abnormal hemoglobins found in Thailand. Southeast Asian J Trop Med Public Health. 1999;30(Suppl. 2):88–93. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous