

Global diversity and inferred ecophysiology of microorganisms with the potential for dissimilatory sulfate/sulfite reduction
- PMID: 37796897
- PMCID: PMC10591310
- DOI: 10.1093/femsre/fuad058
Global diversity and inferred ecophysiology of microorganisms with the potential for dissimilatory sulfate/sulfite reduction
Abstract
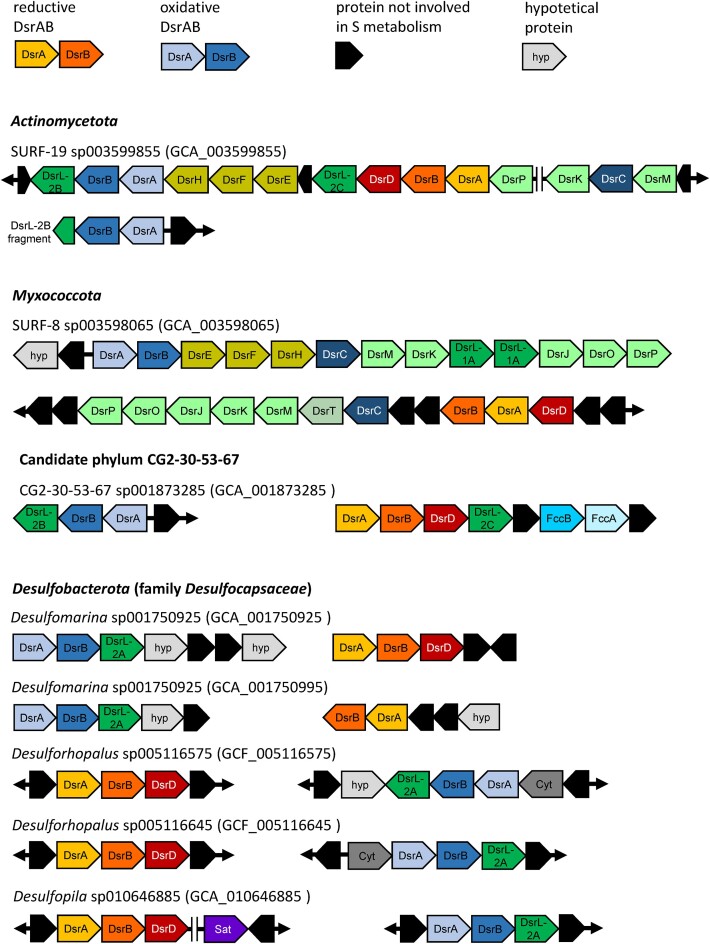
Sulfate/sulfite-reducing microorganisms (SRM) are ubiquitous in nature, driving the global sulfur cycle. A hallmark of SRM is the dissimilatory sulfite reductase encoded by the genes dsrAB. Based on analysis of 950 mainly metagenome-derived dsrAB-carrying genomes, we redefine the global diversity of microorganisms with the potential for dissimilatory sulfate/sulfite reduction and uncover genetic repertoires that challenge earlier generalizations regarding their mode of energy metabolism. We show: (i) 19 out of 23 bacterial and 2 out of 4 archaeal phyla harbor uncharacterized SRM, (ii) four phyla including the Desulfobacterota harbor microorganisms with the genetic potential to switch between sulfate/sulfite reduction and sulfur oxidation, and (iii) the combination as well as presence/absence of different dsrAB-types, dsrL-types and dsrD provides guidance on the inferred direction of dissimilatory sulfur metabolism. We further provide an updated dsrAB database including > 60% taxonomically resolved, uncultured family-level lineages and recommendations on existing dsrAB-targeted primers for environmental surveys. Our work summarizes insights into the inferred ecophysiology of newly discovered SRM, puts SRM diversity into context of the major recent changes in bacterial and archaeal taxonomy, and provides an up-to-date framework to study SRM in a global context.
Keywords: dsrAB; dissimilatory sulfite reductase; metagenomics; sulfate reduction; sulfur cycle; sulfur oxidation.
© The Author(s) 2023. Published by Oxford University Press on behalf of FEMS.
Conflict of interest statement
None declared.
Figures






References
-
- Arshad A, Dalcin Martins P, Frank Jet al. Mimicking microbial interactions under nitrate-reducing conditions in an anoxic bioreactor: enrichment of novel Nitrospirae bacteria distantly related to Thermodesulfovibrio. Environ Microbiol. 2017;19:4965–77. - PubMed
-
- Barton LL, Ritz NL, Fauque GDet al. Sulfur cycling and the intestinal microbiome. Dig Dis Sci. 2017;62:2241–57. - PubMed
-
- Boetius A, Ravenschlag K, Schubert CJet al. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature. 2000;407:623–6. - PubMed
