

Mitophagy for cardioprotection
- PMID: 37798455
- PMCID: PMC10556134
- DOI: 10.1007/s00395-023-01009-x
Mitophagy for cardioprotection
Abstract
Mitochondrial function is maintained by several strictly coordinated mechanisms, collectively termed mitochondrial quality control mechanisms, including fusion and fission, degradation, and biogenesis. As the primary source of energy in cardiomyocytes, mitochondria are the central organelle for maintaining cardiac function. Since adult cardiomyocytes in humans rarely divide, the number of dysfunctional mitochondria cannot easily be diluted through cell division. Thus, efficient degradation of dysfunctional mitochondria is crucial to maintaining cellular function. Mitophagy, a mitochondria specific form of autophagy, is a major mechanism by which damaged or unnecessary mitochondria are targeted and eliminated. Mitophagy is active in cardiomyocytes at baseline and in response to stress, and plays an essential role in maintaining the quality of mitochondria in cardiomyocytes. Mitophagy is mediated through multiple mechanisms in the heart, and each of these mechanisms can partially compensate for the loss of another mechanism. However, insufficient levels of mitophagy eventually lead to mitochondrial dysfunction and the development of heart failure. In this review, we discuss the molecular mechanisms of mitophagy in the heart and the role of mitophagy in cardiac pathophysiology, with the focus on recent findings in the field.
Keywords: Alternative mitophagy; Drp1; Mitochondrial quality control; Mitophagy.
© 2023. Springer-Verlag GmbH Germany, part of Springer Nature.
Conflict of interest statement
The authors declare no coflict of interests.
Figures
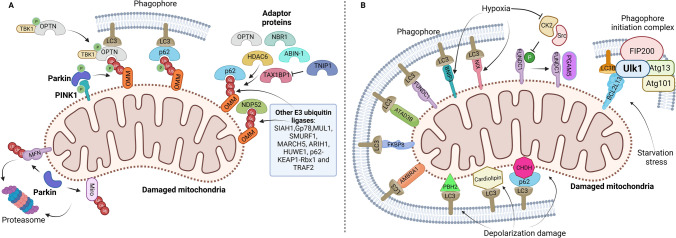
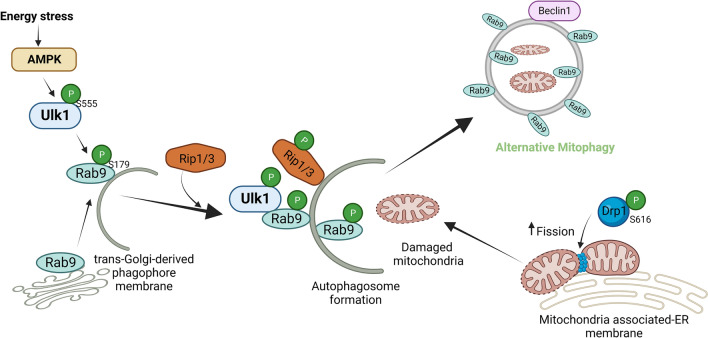
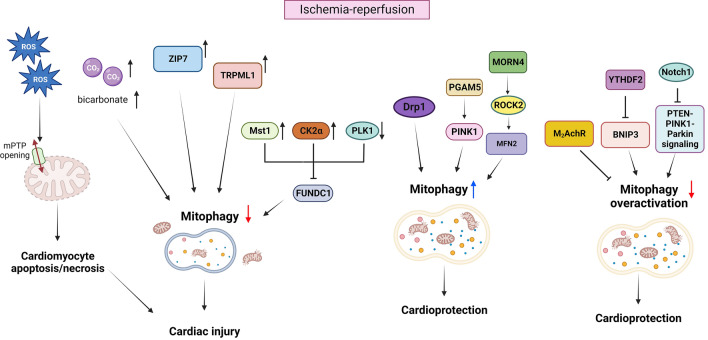

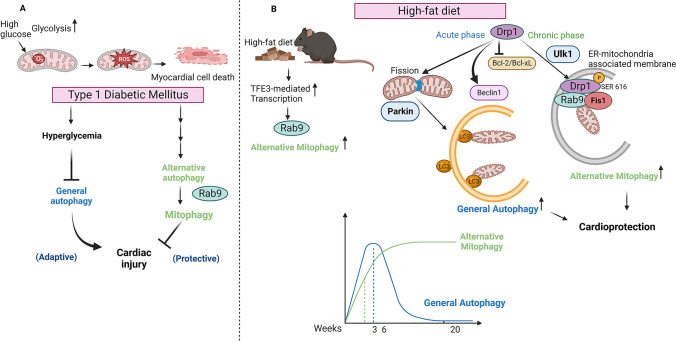
References
-
- Acin-Perez R, Lechuga-Vieco AV, Del Mar MM, Nieto-Arellano R, Torroja C, Sánchez-Cabo F, Jiménez C, González-Guerra A, Carrascoso I, Benincá C, Quiros PM, López-Otín C, Castellano JM, Ruíz-Cabello J, Jiménez-Borreguero LJ, Enríquez JA. Ablation of the stress protease OMA1 protects against heart failure in mice. Sci Transl Med. 2018;10:eaan4935. doi: 10.1126/scitranslmed.aan4935. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
