

GET_PANGENES: calling pangenes from plant genome alignments confirms presence-absence variation
- PMID: 37798615
- PMCID: PMC10552430
- DOI: 10.1186/s13059-023-03071-z
GET_PANGENES: calling pangenes from plant genome alignments confirms presence-absence variation
Abstract
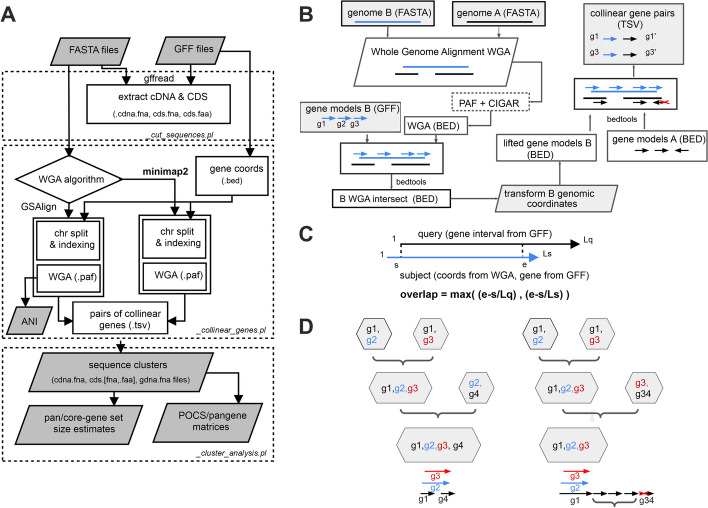
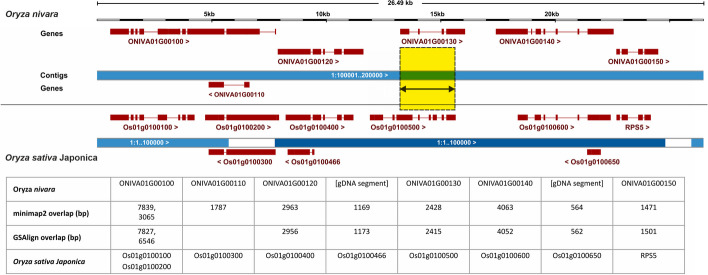
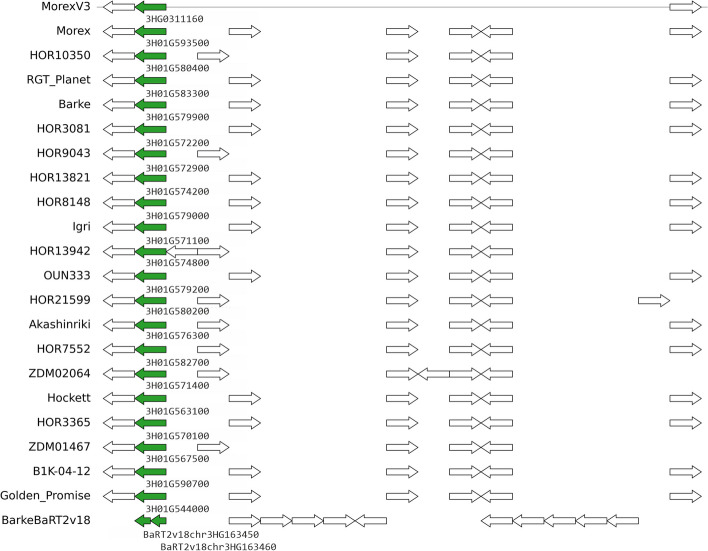
Crop pangenomes made from individual cultivar assemblies promise easy access to conserved genes, but genome content variability and inconsistent identifiers hamper their exploration. To address this, we define pangenes, which summarize a species coding potential and link back to original annotations. The protocol get_pangenes performs whole genome alignments (WGA) to call syntenic gene models based on coordinate overlaps. A benchmark with small and large plant genomes shows that pangenes recapitulate phylogeny-based orthologies and produce complete soft-core gene sets. Moreover, WGAs support lift-over and help confirm gene presence-absence variation. Source code and documentation: https://github.com/Ensembl/plant-scripts .
Keywords: Collinearity; Gene annotation; Pangene; Plant genome; Presence-absence variation; Whole genome alignment.
© 2023. BioMed Central Ltd., part of Springer Nature.
Conflict of interest statement
Paul Flicek is a member of the Scientific Advisory Boards of Fabric Genomics, Inc. and Eagle Genomics, Ltd.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
