

Expanding a precision medicine platform for malignant peripheral nerve sheath tumors: New patient-derived orthotopic xenografts, cell lines and tumor entities
- PMID: 37798904
- PMCID: PMC10994238
- DOI: 10.1002/1878-0261.13534
Expanding a precision medicine platform for malignant peripheral nerve sheath tumors: New patient-derived orthotopic xenografts, cell lines and tumor entities
Abstract
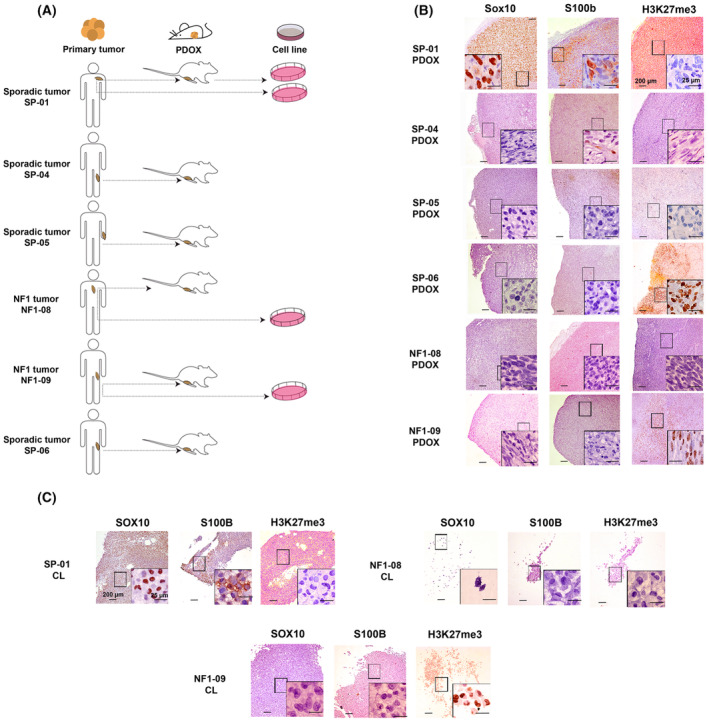
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive soft-tissue sarcomas with a poor survival rate, presenting either sporadically or in the context of neurofibromatosis type 1 (NF1). The histological diagnosis of MPNSTs can be challenging, with different tumors exhibiting great histological and marker expression overlap. This heterogeneity could be partly responsible for the observed disparity in treatment response due to the inherent diversity of the preclinical models used. For several years, our group has been generating a large patient-derived orthotopic xenograft (PDOX) MPNST platform for identifying new precision medicine treatments. Herein, we describe the expansion of this platform using six primary tumors clinically diagnosed as MPNSTs, from which we obtained six additional PDOX mouse models and three cell lines, thus generating three pairs of in vitro-in vivo models. We extensively characterized these tumors and derived preclinical models, including genomic, epigenomic, and histological analyses. Tumors were reclassified after these analyses: three remained as MPNSTs (two being classic MPNSTs), one was a melanoma, another was a neurotrophic tyrosine receptor kinase (NTRK)-rearranged spindle cell neoplasm, and, finally, the last was an unclassifiable tumor bearing neurofibromin-2 (NF2) inactivation, a neuroblastoma RAS viral oncogene homolog (NRAS) oncogenic mutation, and a SWI/SNF-related matrix-associated actin-dependent regulator of chromatin (SMARCA4) heterozygous truncated variant. New cell lines and PDOXs faithfully recapitulated histology, marker expression, and genomic characteristics of the primary tumors. The diversity in tumor identity and their specific associated genomic alterations impacted treatment responses obtained when we used the new cell lines for testing compounds against known altered pathways in MPNSTs. In summary, we present here an extension of our MPNST precision medicine platform, with new PDOXs and cell lines, including tumor entities confounded as MPNSTs in a real clinical scenario. This platform may constitute a useful tool for obtaining correct preclinical information to guide MPNST clinical trials.
Keywords: MPNST; NF1; PDOX; cellular models; treatment response; tumor entities.
© 2023 The Authors. Molecular Oncology published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




Similar articles
-
Triple Combination of MEK, BET, and CDK Inhibitors Significantly Reduces Human Malignant Peripheral Nerve Sheath Tumors in Mouse Models.Clin Cancer Res. 2025 Mar 3;31(5):907-920. doi: 10.1158/1078-0432.CCR-24-2807. Clin Cancer Res. 2025. PMID: 39786423 Free PMC article.
-
Malignant peripheral nerve sheath tumor (MPNST) and MPNST-like entities are defined by a specific DNA methylation profile in pediatric and juvenile population.Clin Epigenetics. 2024 Jan 4;16(1):9. doi: 10.1186/s13148-023-01621-7. Clin Epigenetics. 2024. PMID: 38178234 Free PMC article.
-
Comprehensive establishment and characterization of orthoxenograft mouse models of malignant peripheral nerve sheath tumors for personalized medicine.EMBO Mol Med. 2015 May;7(5):608-27. doi: 10.15252/emmm.201404430. EMBO Mol Med. 2015. PMID: 25810463 Free PMC article.
-
New Model Systems and the Development of Targeted Therapies for the Treatment of Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors.Genes (Basel). 2020 Apr 28;11(5):477. doi: 10.3390/genes11050477. Genes (Basel). 2020. PMID: 32353955 Free PMC article. Review.
-
Malignant peripheral nerve sheath tumor: models, biology, and translation.Oncogene. 2022 Apr;41(17):2405-2421. doi: 10.1038/s41388-022-02290-1. Epub 2022 Apr 7. Oncogene. 2022. PMID: 35393544 Free PMC article. Review.
Cited by
-
Triple Combination of MEK, BET, and CDK Inhibitors Significantly Reduces Human Malignant Peripheral Nerve Sheath Tumors in Mouse Models.Clin Cancer Res. 2025 Mar 3;31(5):907-920. doi: 10.1158/1078-0432.CCR-24-2807. Clin Cancer Res. 2025. PMID: 39786423 Free PMC article.
-
New models for MPNST: establishment and comprehensive characterization of two tumor cell lines.Cancer Cell Int. 2025 Jul 18;25(1):268. doi: 10.1186/s12935-025-03845-4. Cancer Cell Int. 2025. PMID: 40682039 Free PMC article.
-
Proactive process evaluation of precision medicine platforms: a roadmap.BMJ Health Care Inform. 2025 Jul 20;32(1):e101434. doi: 10.1136/bmjhci-2025-101434. BMJ Health Care Inform. 2025. PMID: 40685163 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
