

Modeling of horizontal pleiotropy identifies possible causal gene expression in systemic lupus erythematosus
- PMID: 37799268
- PMCID: PMC10554754
- DOI: 10.3389/flupu.2023.1234578
Modeling of horizontal pleiotropy identifies possible causal gene expression in systemic lupus erythematosus
Abstract
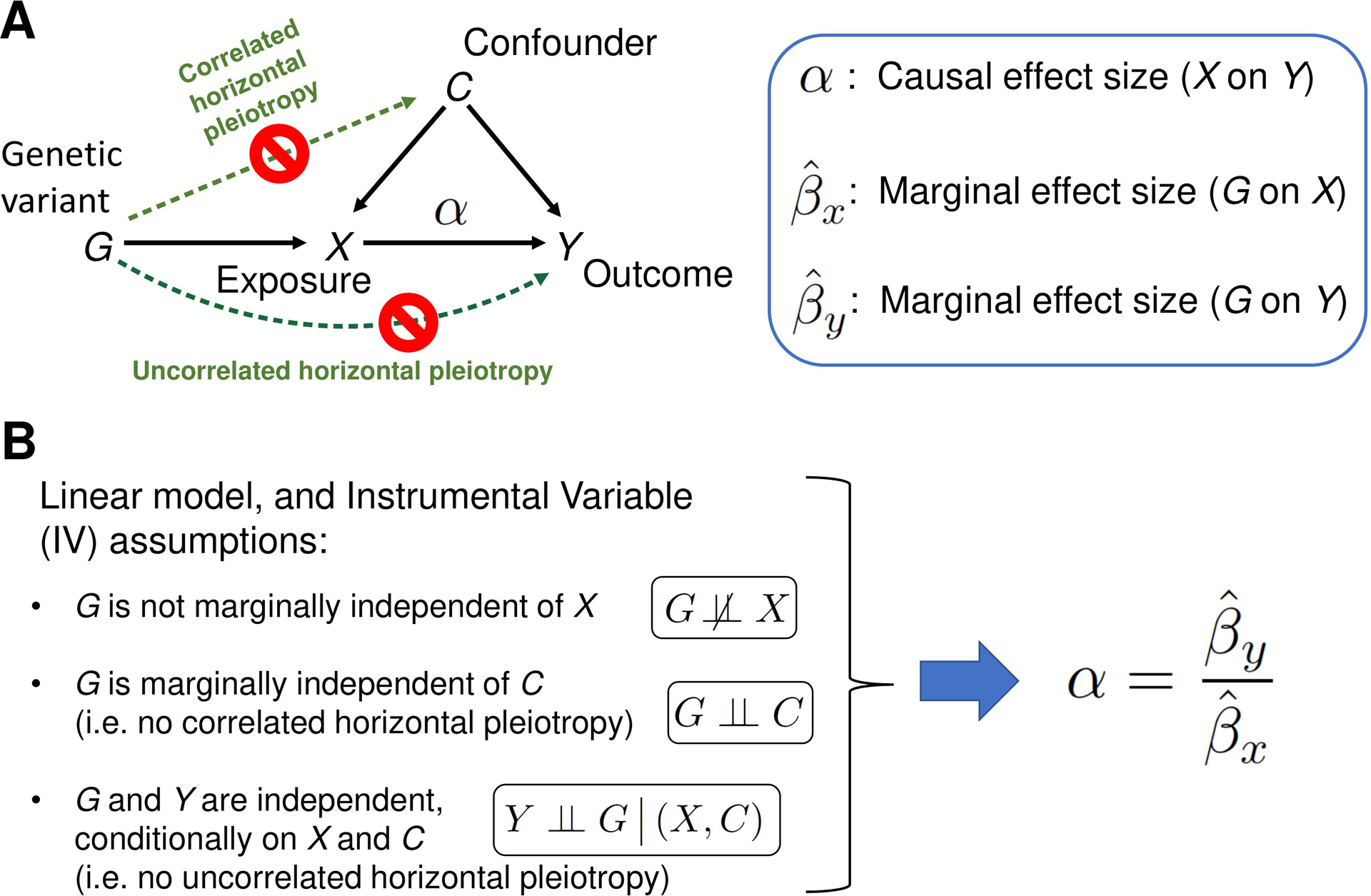
Background: Systemic lupus erythematosus (SLE) is a chronic autoimmune condition with complex causes involving genetic and environmental factors. While genome-wide association studies (GWASs) have identified genetic loci associated with SLE, the functional genomic elements responsible for disease development remain largely unknown. Mendelian Randomization (MR) is an instrumental variable approach to causal inference based on data from observational studies, where genetic variants are employed as instrumental variables (IVs).
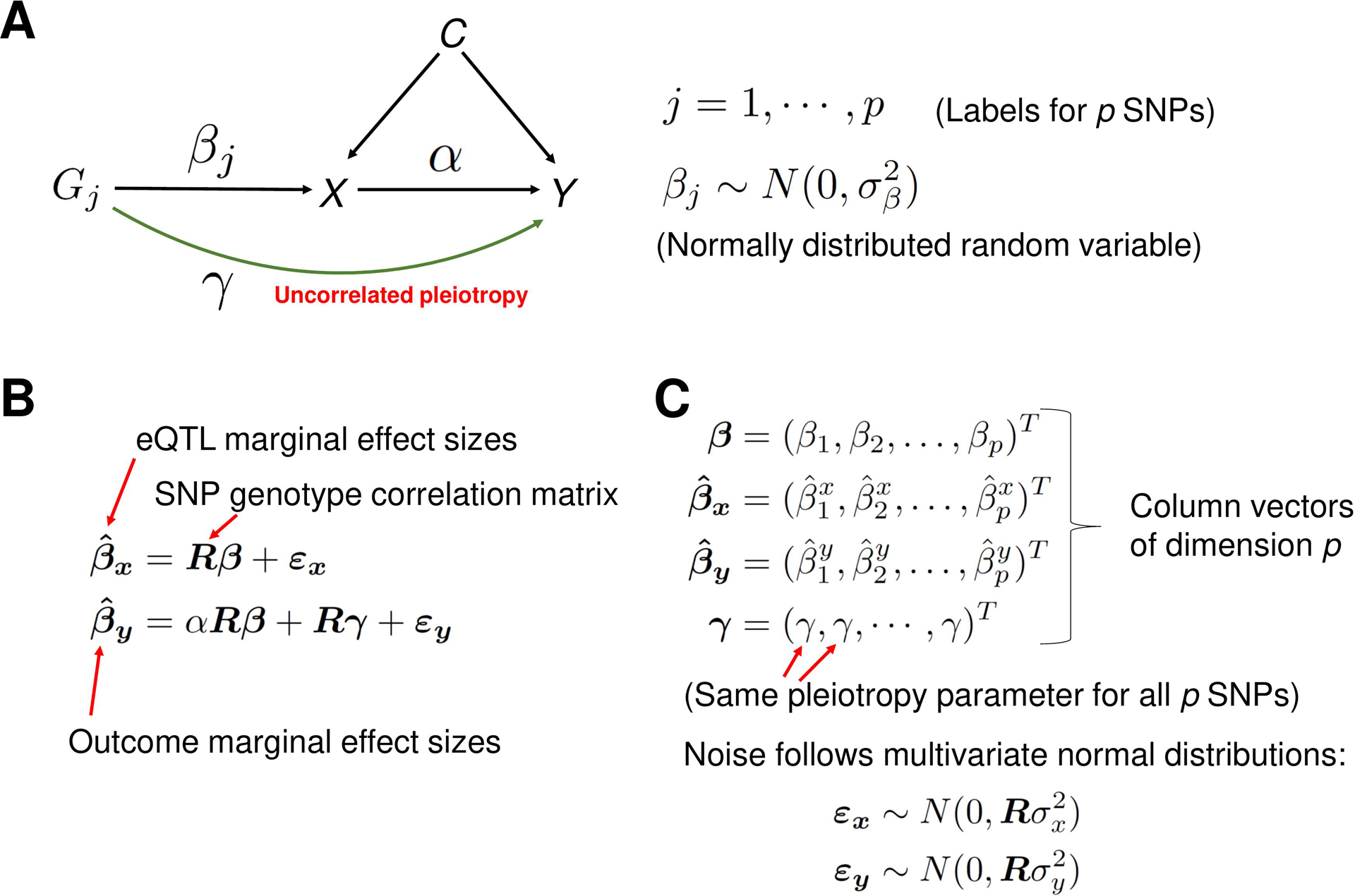
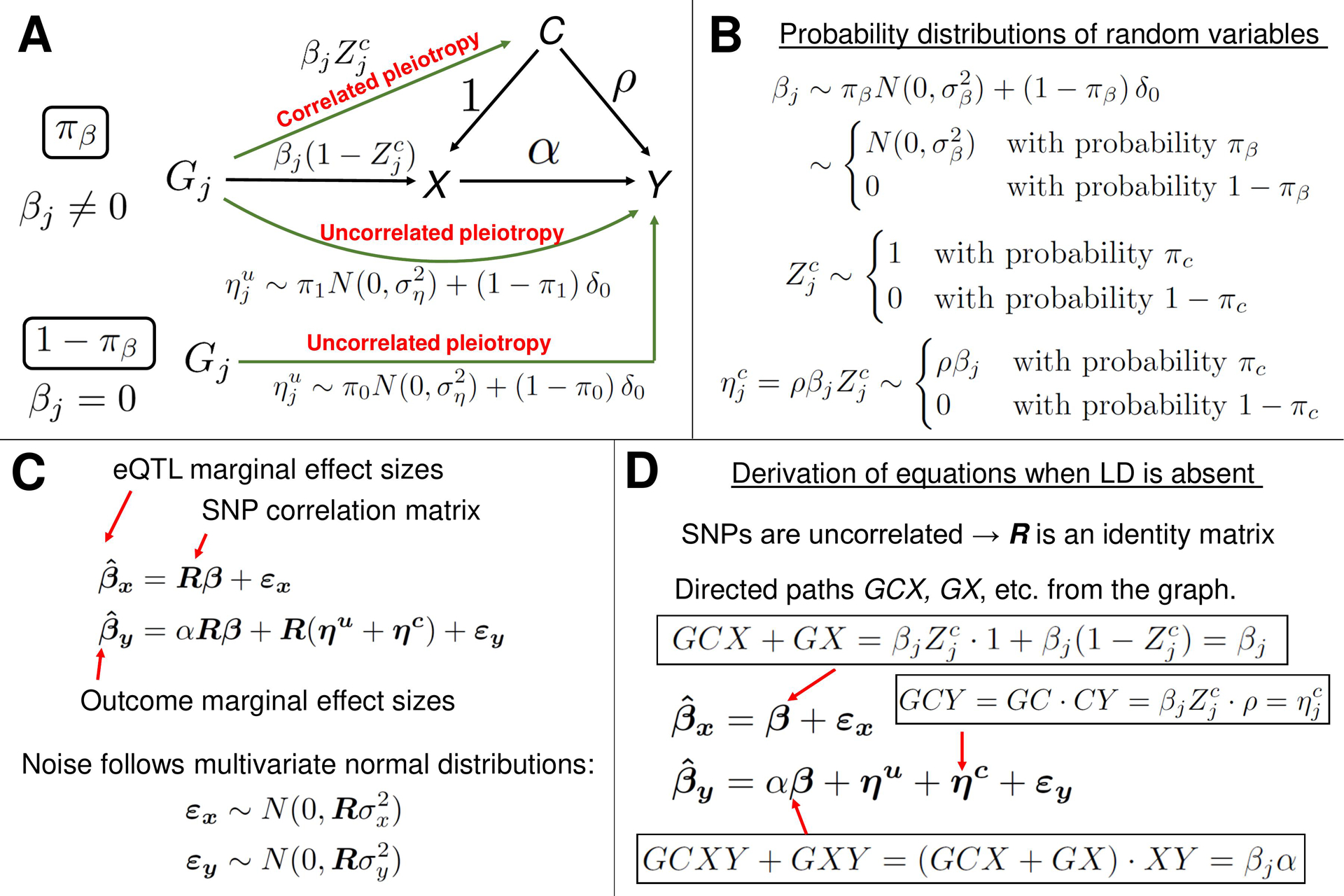
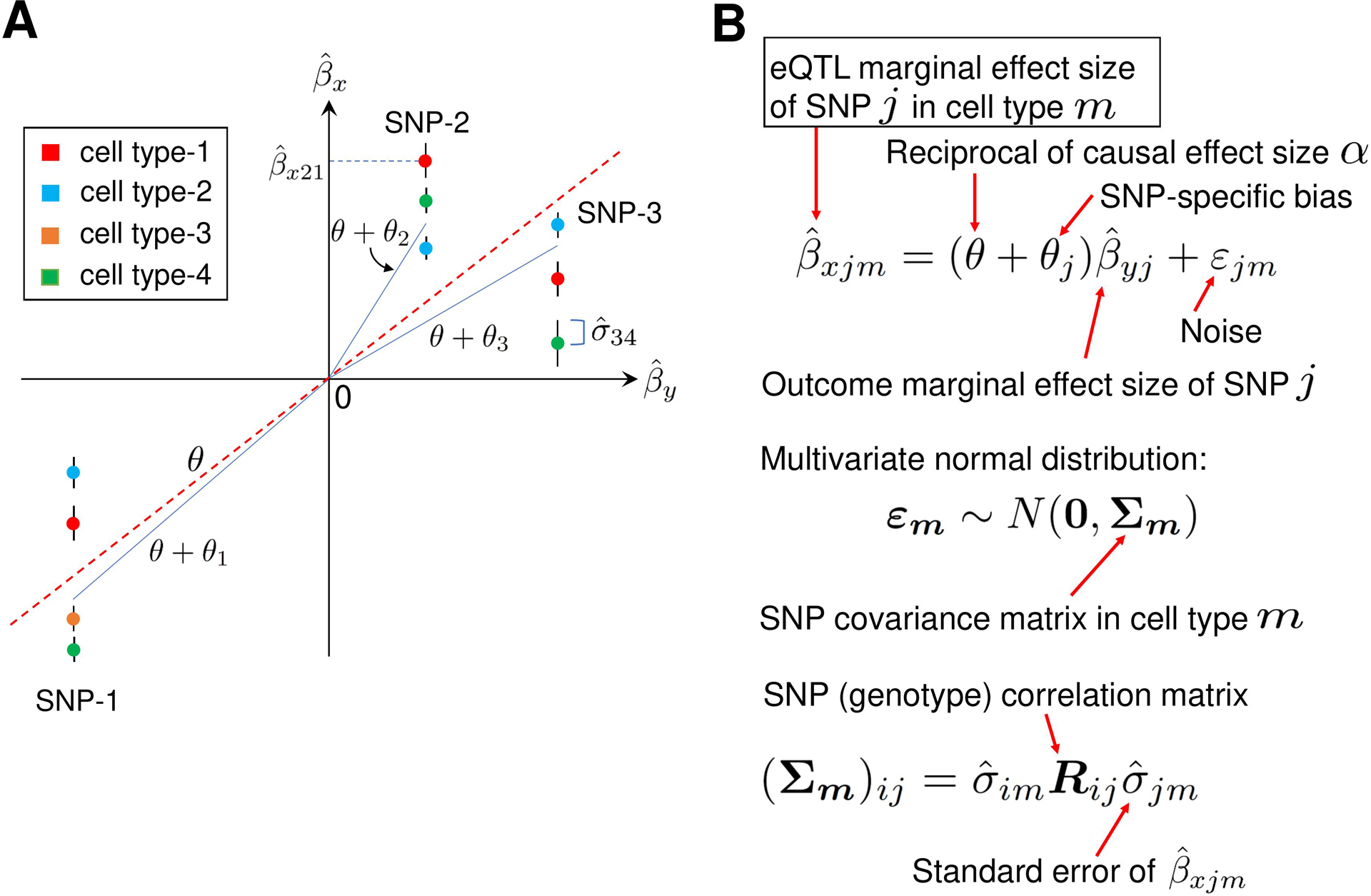
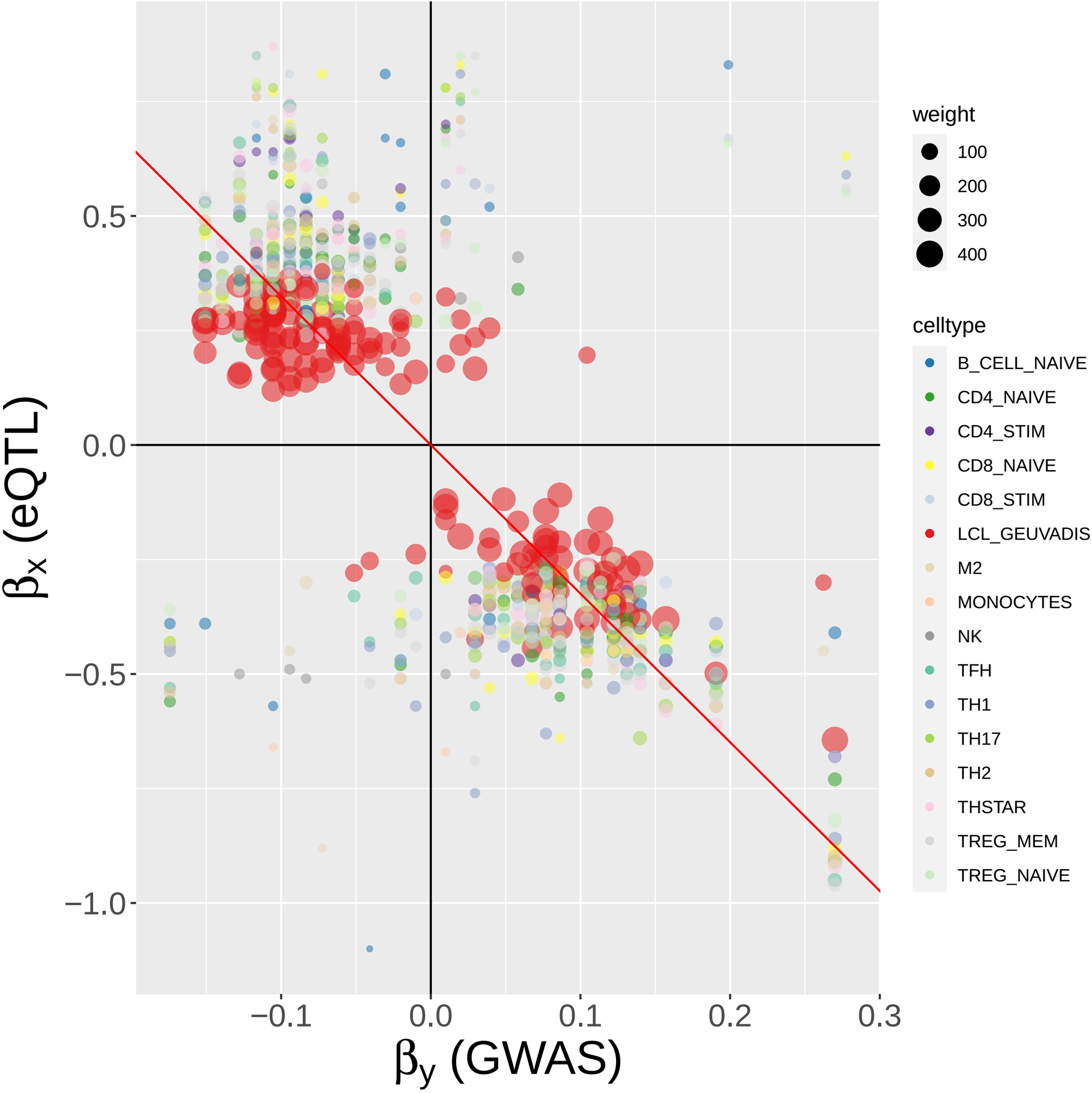
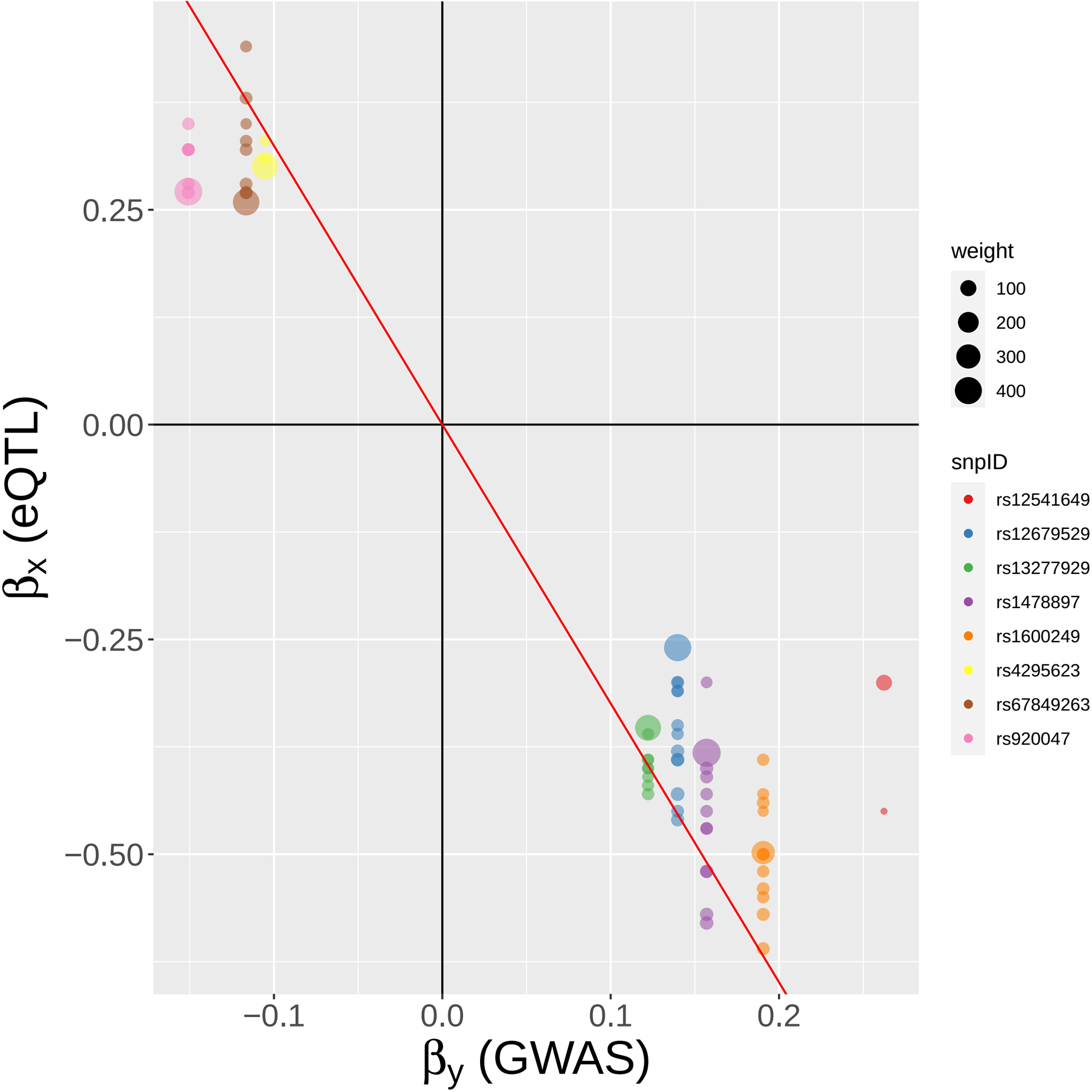
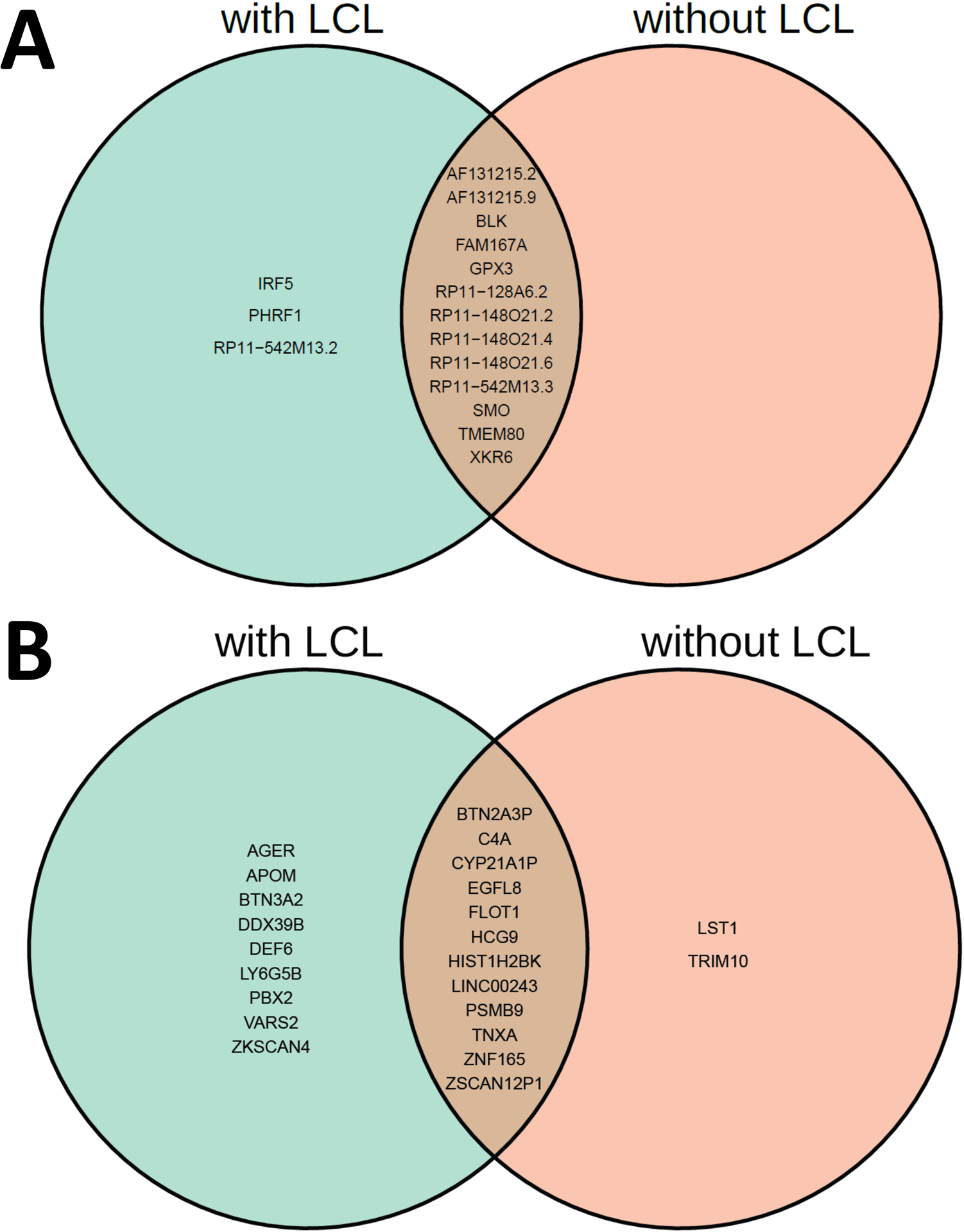
Methods: This study utilized a two-step strategy to identify causal genes for SLE. In the first step, the classical MR method was employed, assuming the absence of horizontal pleiotropy, to estimate the causal effect of gene expression on SLE. In the second step, advanced probabilistic MR methods (PMR-Egger, MRAID, and MR-MtRobin) were applied to the genes identified in the first step, considering horizontal pleiotropy, to filter out false positives. PMR-Egger and MRAID analyses utilized whole blood expression quantitative trait loci (eQTL) and SLE GWAS summary data, while MR-MtRobin analysis used an independent eQTL dataset from multiple immune cell types along with the same SLE GWAS data.
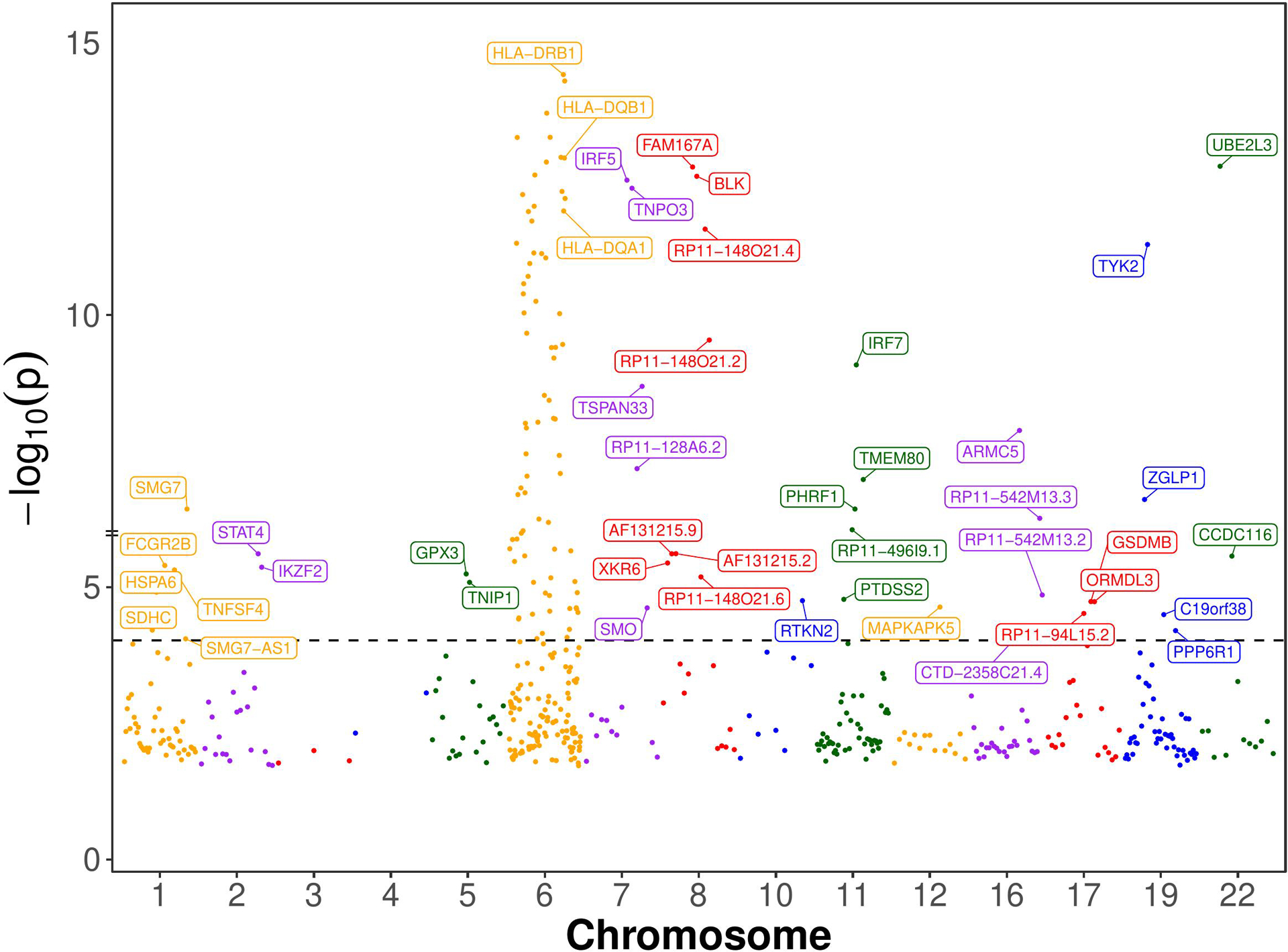
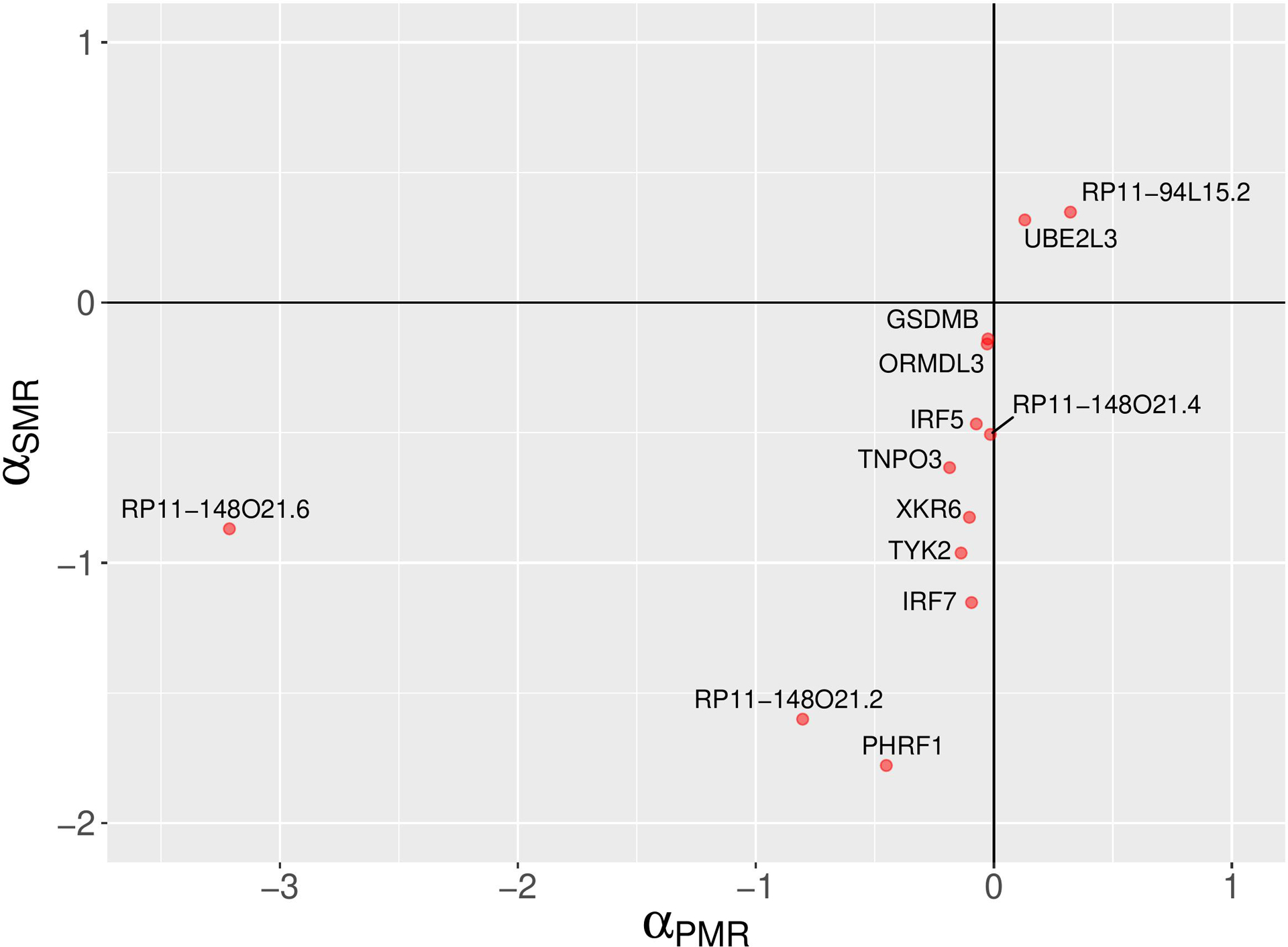
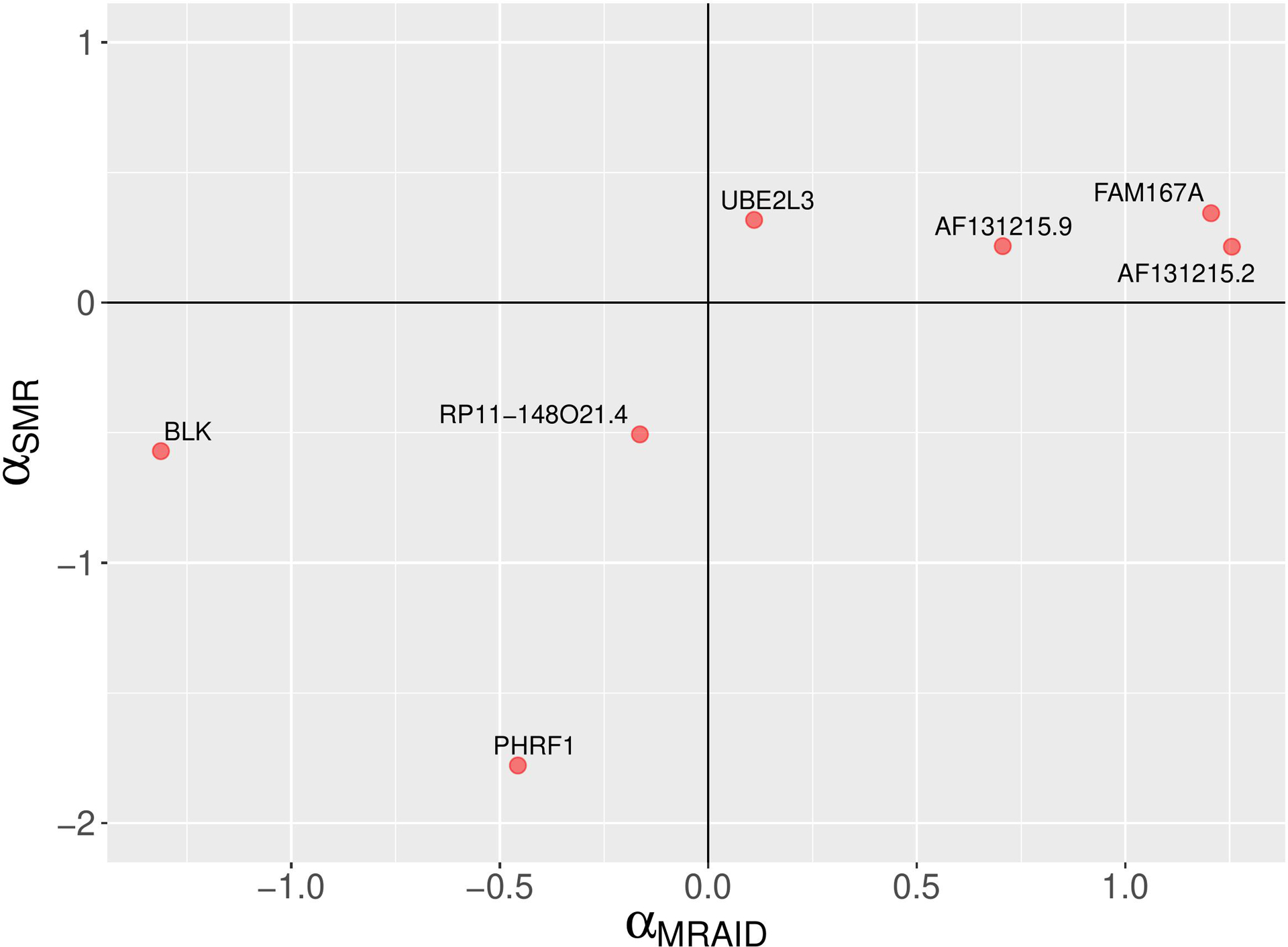
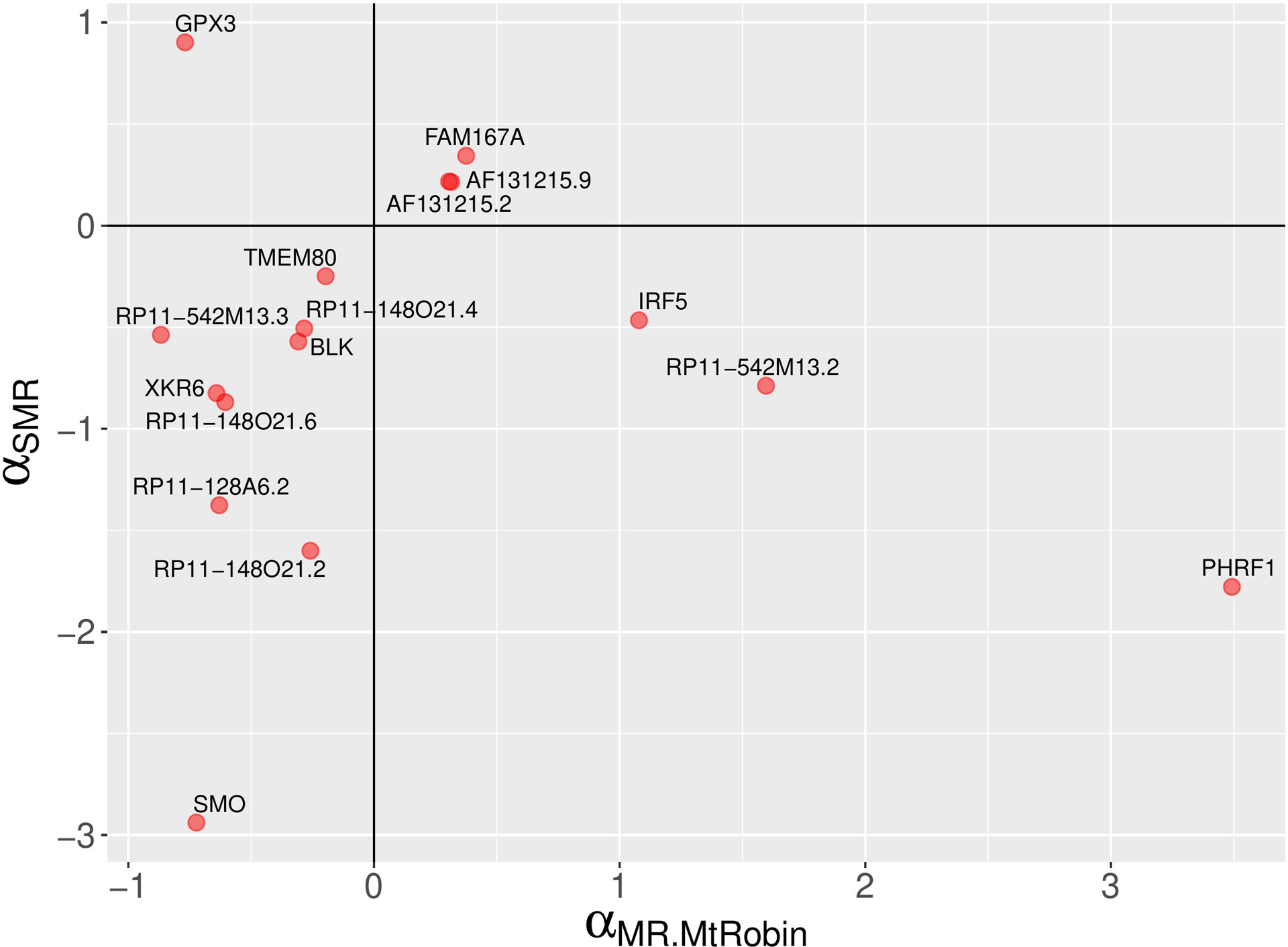
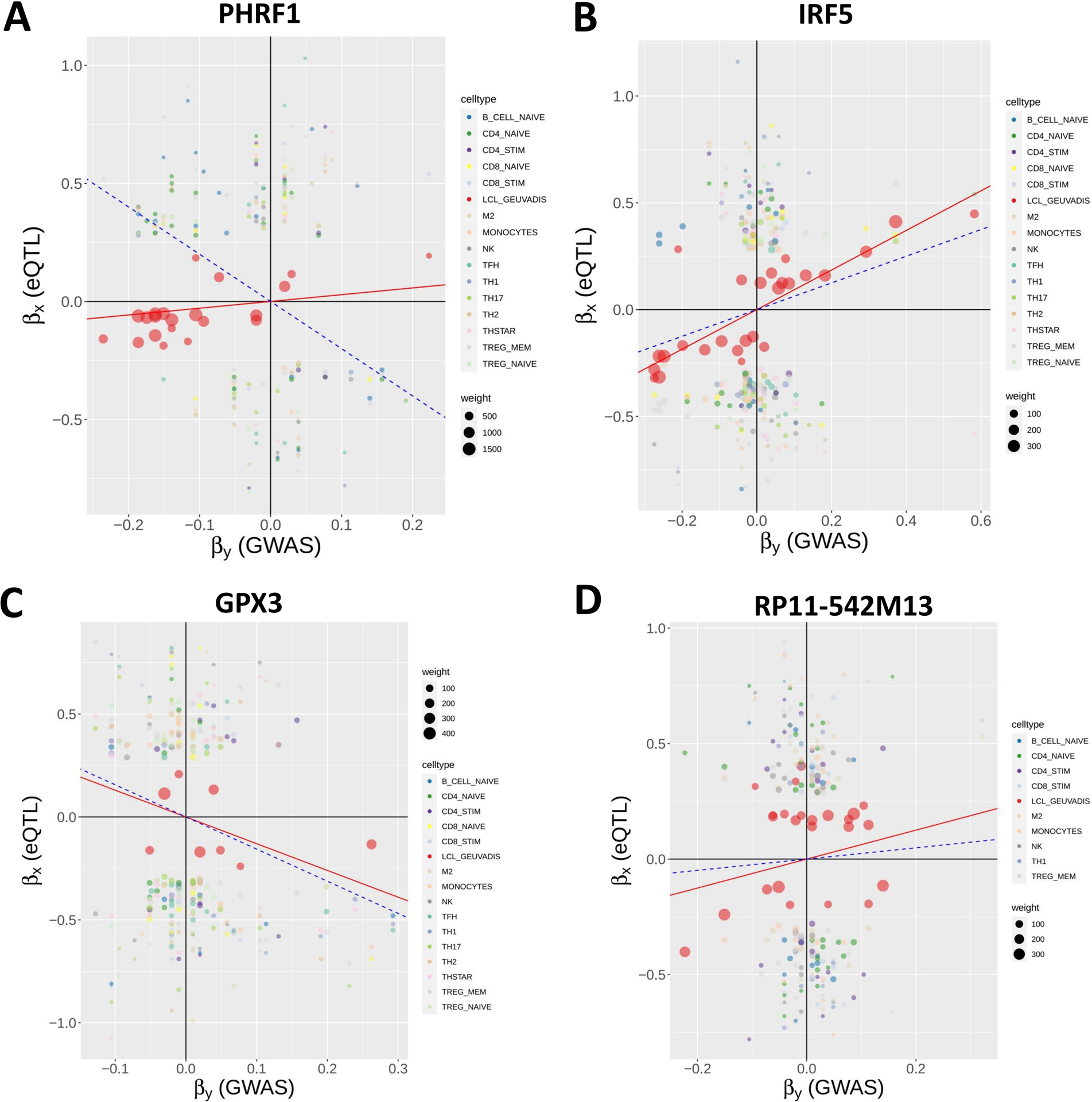
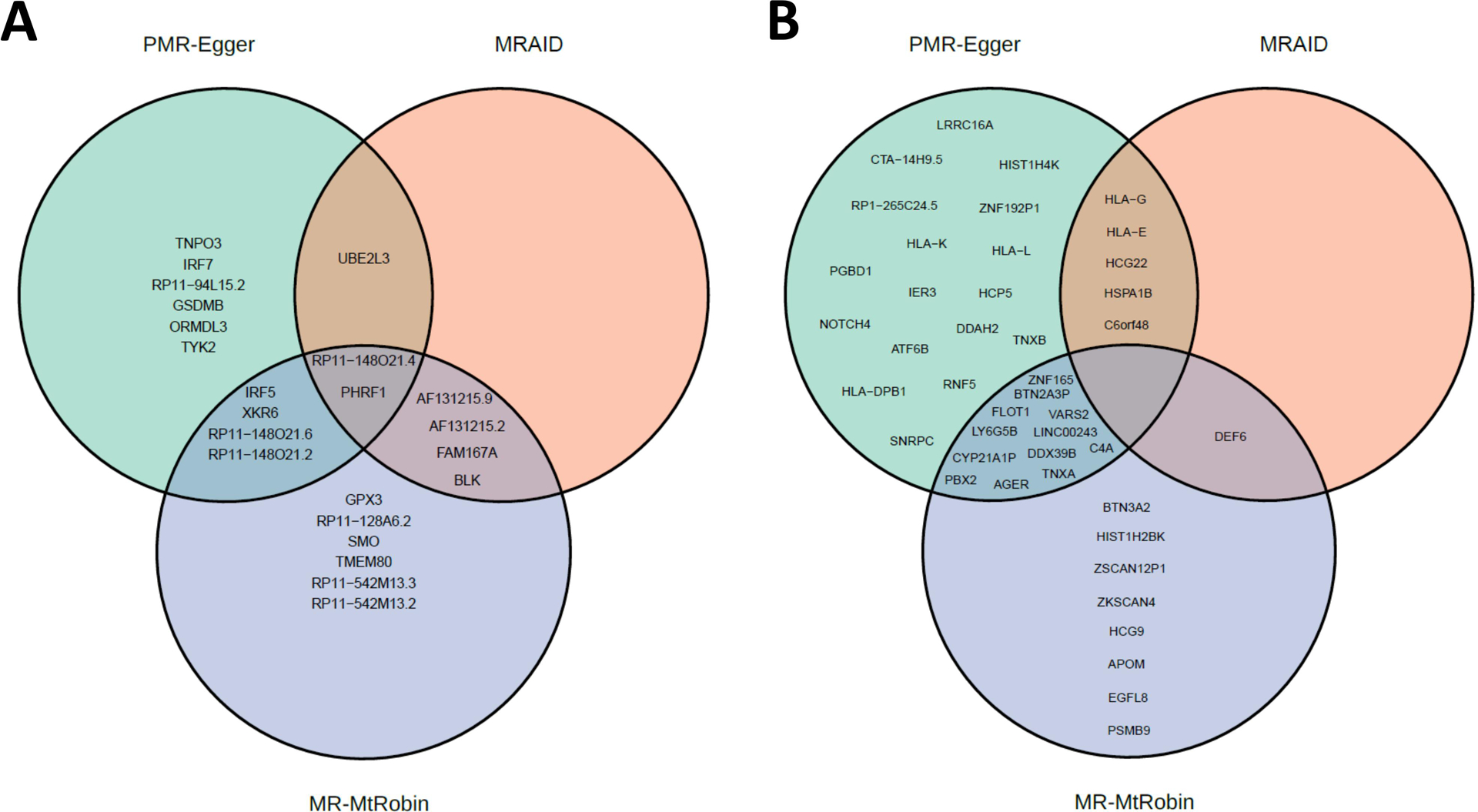
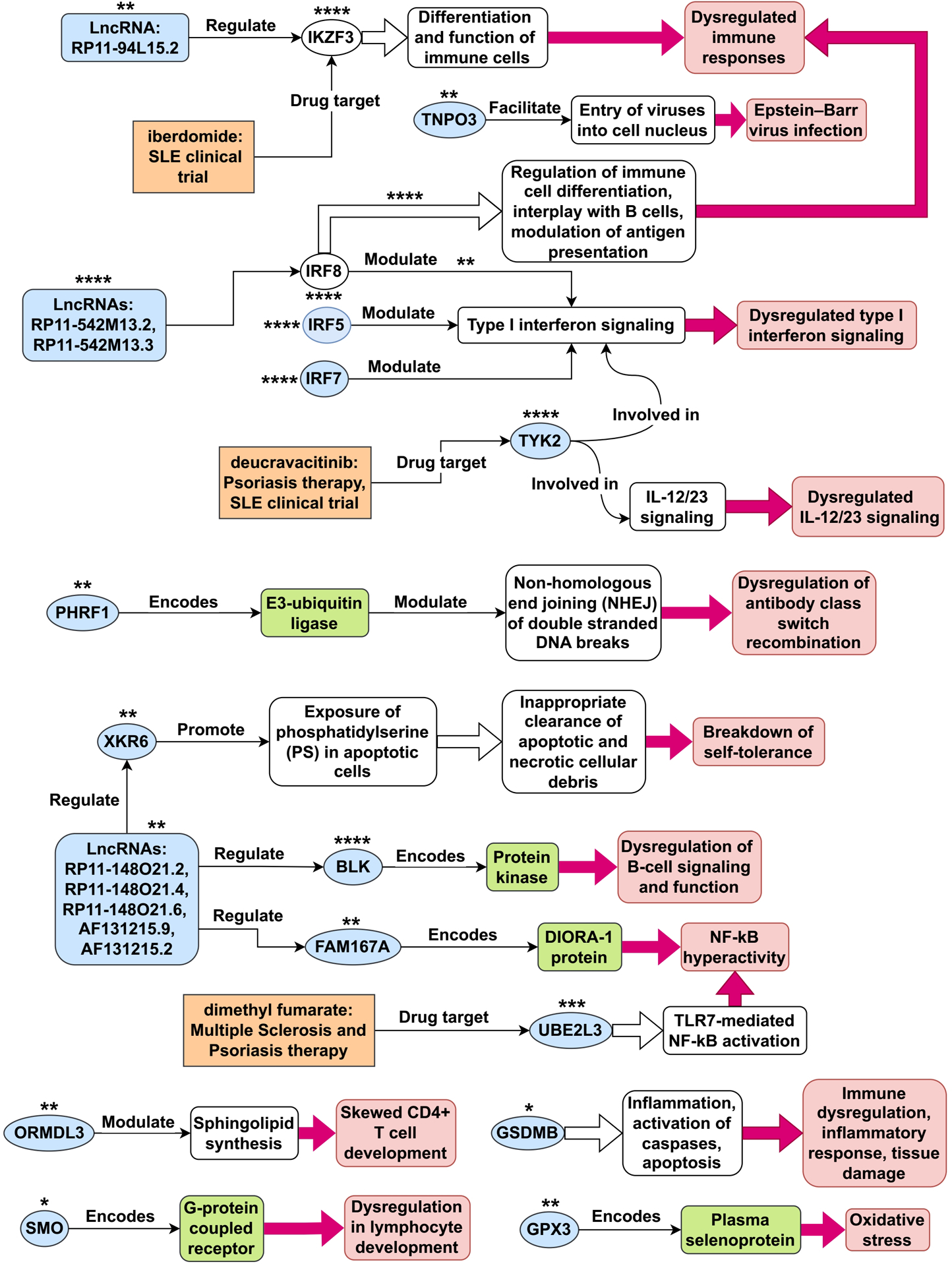
Results: The initial MR analysis identified 142 genes, including 43 outside of chromosome 6. Subsequently, applying the advanced MR methods reduced the number of genes with significant causal effects on SLE to 66. PMR-Egger, MRAID, and MR-MtRobin, respectively, identified 13, 7, and 16 non-chromosome 6 genes with significant causal effects. All methods identified expression of PHRF1 gene as causal for SLE. A comprehensive literature review was conducted to enhance understanding of the functional roles and mechanisms of the identified genes in SLE development.
Conclusions: The findings from the three MR methods exhibited overlapping genes with causal effects on SLE, demonstrating consistent results. However, each method also uncovered unique genes due to different modelling assumptions and technical factors, highlighting the complementary nature of the approaches. Importantly, MRAID demonstrated a reduced percentage of causal genes from the Major Histocompatibility complex (MHC) region on chromosome 6, indicating its potential in minimizing false positive findings. This study contributes to unraveling the mechanisms underlying SLE by employing advanced probabilistic MR methods to identify causal genes, thereby enhancing our understanding of SLE pathogenesis.
Keywords: GWAS; Mendelian Randomization (MR); PHRF1; causal inference; eQTL; gene expression; genetics; systemic lupus erythematosus (SLE).
Figures
Similar articles
-
Association between systemic lupus erythematosus and osteoporosis: a mendelian randomization analysis.BMC Rheumatol. 2024 May 6;8(1):17. doi: 10.1186/s41927-024-00388-5. BMC Rheumatol. 2024. PMID: 38711124 Free PMC article.
-
Systemic lupus erythematosus Association between Osteomyelitis: A two-sample Mendelian randomization study in European population.Heliyon. 2023 Nov 29;9(12):e22999. doi: 10.1016/j.heliyon.2023.e22999. eCollection 2023 Dec. Heliyon. 2023. PMID: 38125497 Free PMC article.
-
Investigating the causal relationship and potential shared diagnostic genes between primary biliary cholangitis and systemic lupus erythematosus using bidirectional Mendelian randomization and transcriptomic analyses.Front Immunol. 2024 Feb 19;15:1270401. doi: 10.3389/fimmu.2024.1270401. eCollection 2024. Front Immunol. 2024. PMID: 38464525 Free PMC article.
-
A review of genetic risk in systemic lupus erythematosus.Expert Rev Clin Immunol. 2023 Jul-Dec;19(10):1247-1258. doi: 10.1080/1744666X.2023.2240959. Epub 2023 Jul 26. Expert Rev Clin Immunol. 2023. PMID: 37496418 Review.
-
Causal inference and GWAS: Rubin, Pearl, and Mendelian randomization.J Anim Breed Genet. 2025 Mar;142(2):200-213. doi: 10.1111/jbg.12898. Epub 2024 Aug 28. J Anim Breed Genet. 2025. PMID: 39193621 Review.
Cited by
-
Unlocking therapeutic potential: Targeting lymphocyte activation Gene-3 (LAG-3) with fibrinogen-like protein 1 (FGL1) in systemic lupus erythematosus.J Transl Autoimmun. 2024 Jul 27;9:100249. doi: 10.1016/j.jtauto.2024.100249. eCollection 2024 Dec. J Transl Autoimmun. 2024. PMID: 39228513 Free PMC article. Review.
-
Mendelian randomization in cancer research: opportunities and challenges.Infect Agent Cancer. 2025 Jun 15;20(1):37. doi: 10.1186/s13027-025-00672-0. Infect Agent Cancer. 2025. PMID: 40518507 Free PMC article.
-
Pervasive Sharing of Causal Genetic Risk Factors Contributes to Clinical and Molecular Overlap between Sjögren's Disease and Systemic Lupus Erythematosus.Int J Mol Sci. 2023 Sep 22;24(19):14449. doi: 10.3390/ijms241914449. Int J Mol Sci. 2023. PMID: 37833897 Free PMC article.
References
-
- Afrasiabi A, Keane JT, Ong LTC, Alinejad-Rokny H, Fewings NL, Booth DR, Parnell GP, Swaminathan S, 2022. Genetic and transcriptomic analyses support a switch to lytic phase in Epstein Barr virus infection as an important driver in developing Systemic Lupus Erythematosus. J. Autoimmun 127, 102781. 10.1016/j.jaut.2021.102781 - DOI - PubMed
-
- Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, Martín J, Fairfax BP, Knight JC, Chen L, Replogle J, Syvänen A-C, Rönnblom L, Graham RR, Wither JE, Rioux JD, Alarcón-Riquelme ME, Vyse TJ, 2015. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet 47, 1457–1464. 10.1038/ng.3434 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials