

Myocardial Recovery in Recent Onset Dilated Cardiomyopathy: Role of CDCP1 and Cardiac Fibrosis
- PMID: 37800334
- PMCID: PMC10746262
- DOI: 10.1161/CIRCRESAHA.123.323200
Myocardial Recovery in Recent Onset Dilated Cardiomyopathy: Role of CDCP1 and Cardiac Fibrosis
Abstract
Background: Dilated cardiomyopathy (DCM) is a major cause of heart failure and carries a high mortality rate. Myocardial recovery in DCM-related heart failure patients is highly variable, with some patients having little or no response to standard drug therapy. A genome-wide association study may agnostically identify biomarkers and provide novel insight into the biology of myocardial recovery in DCM.
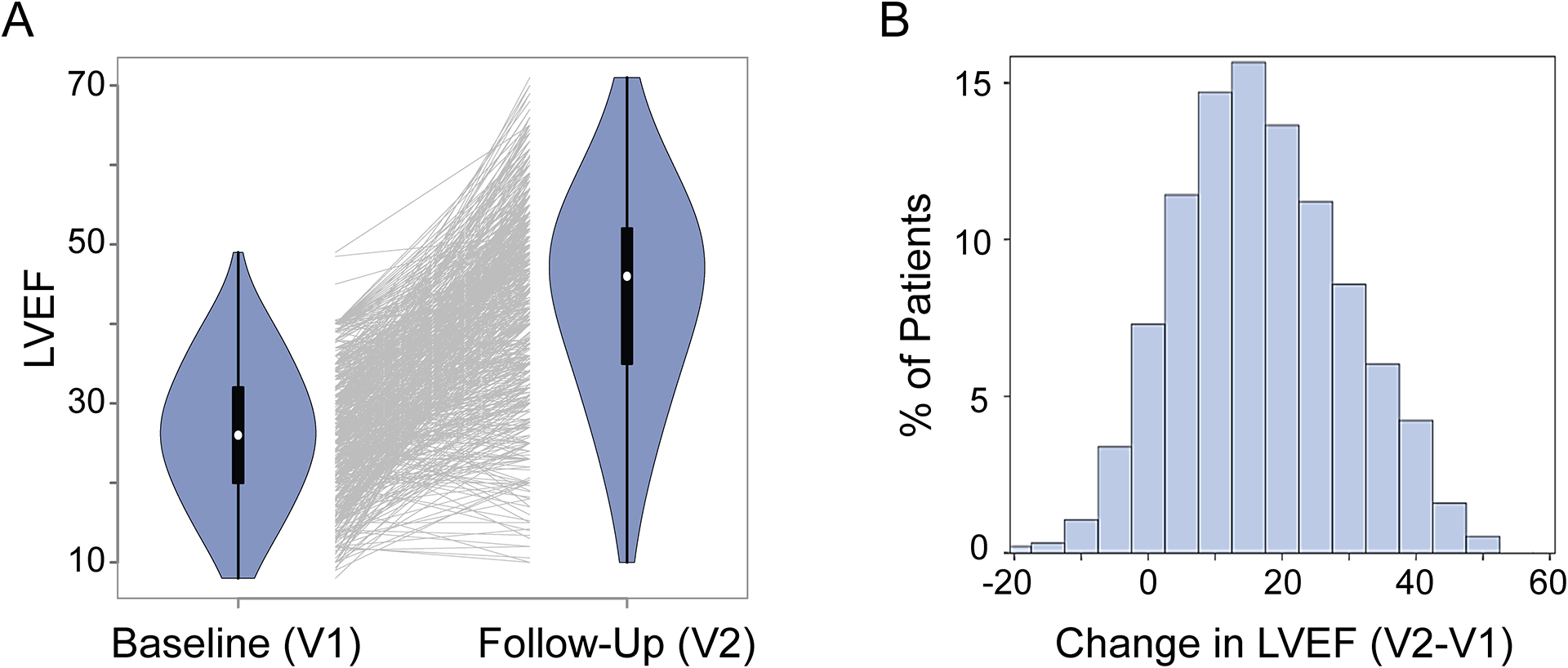
Methods: A genome-wide association study for change in left ventricular ejection fraction was performed in 686 White subjects with recent-onset DCM who received standard pharmacotherapy. Genome-wide association study signals were subsequently functionally validated and studied in relevant cellular models to understand molecular mechanisms that may have contributed to the change in left ventricular ejection fraction.
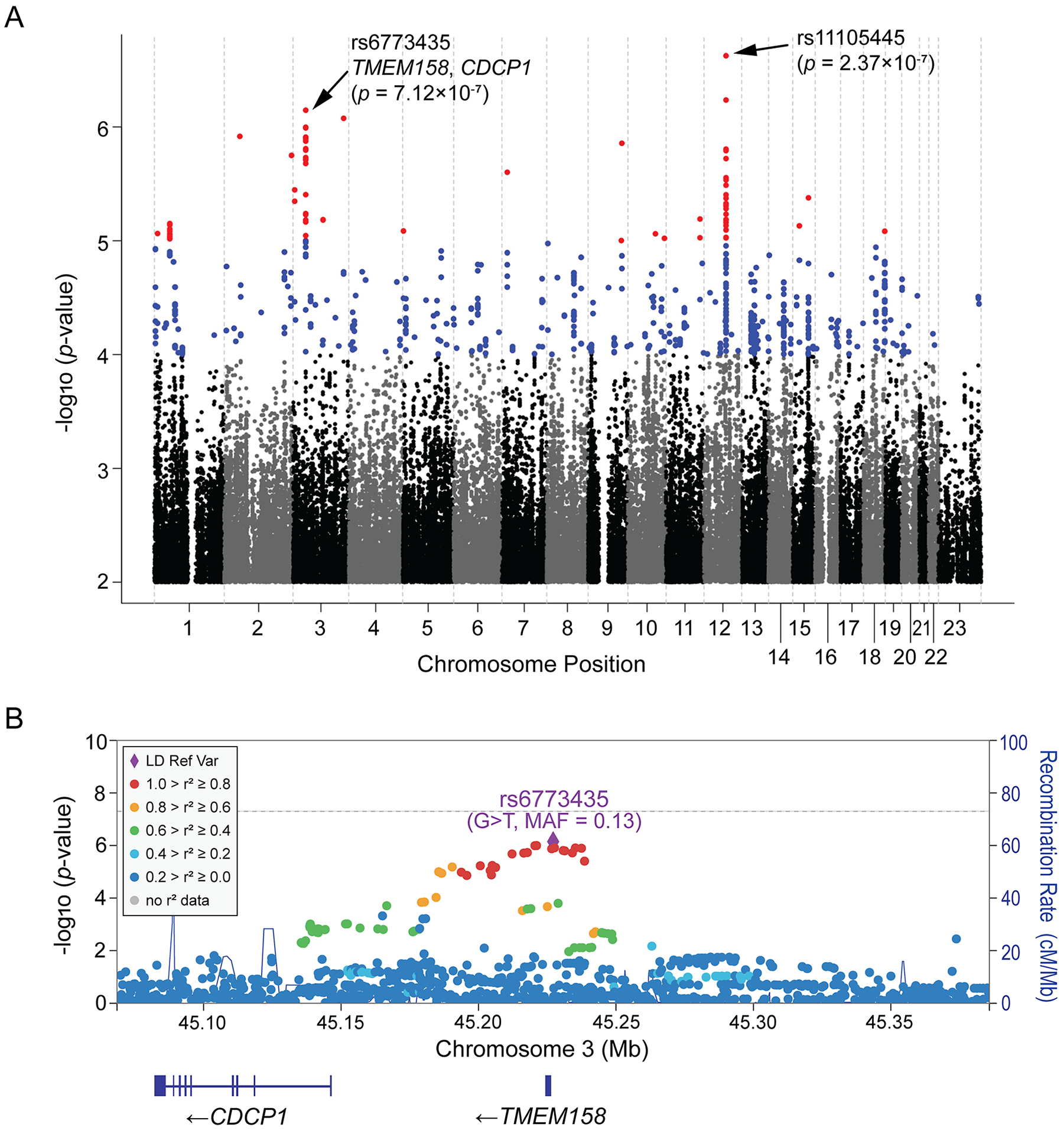
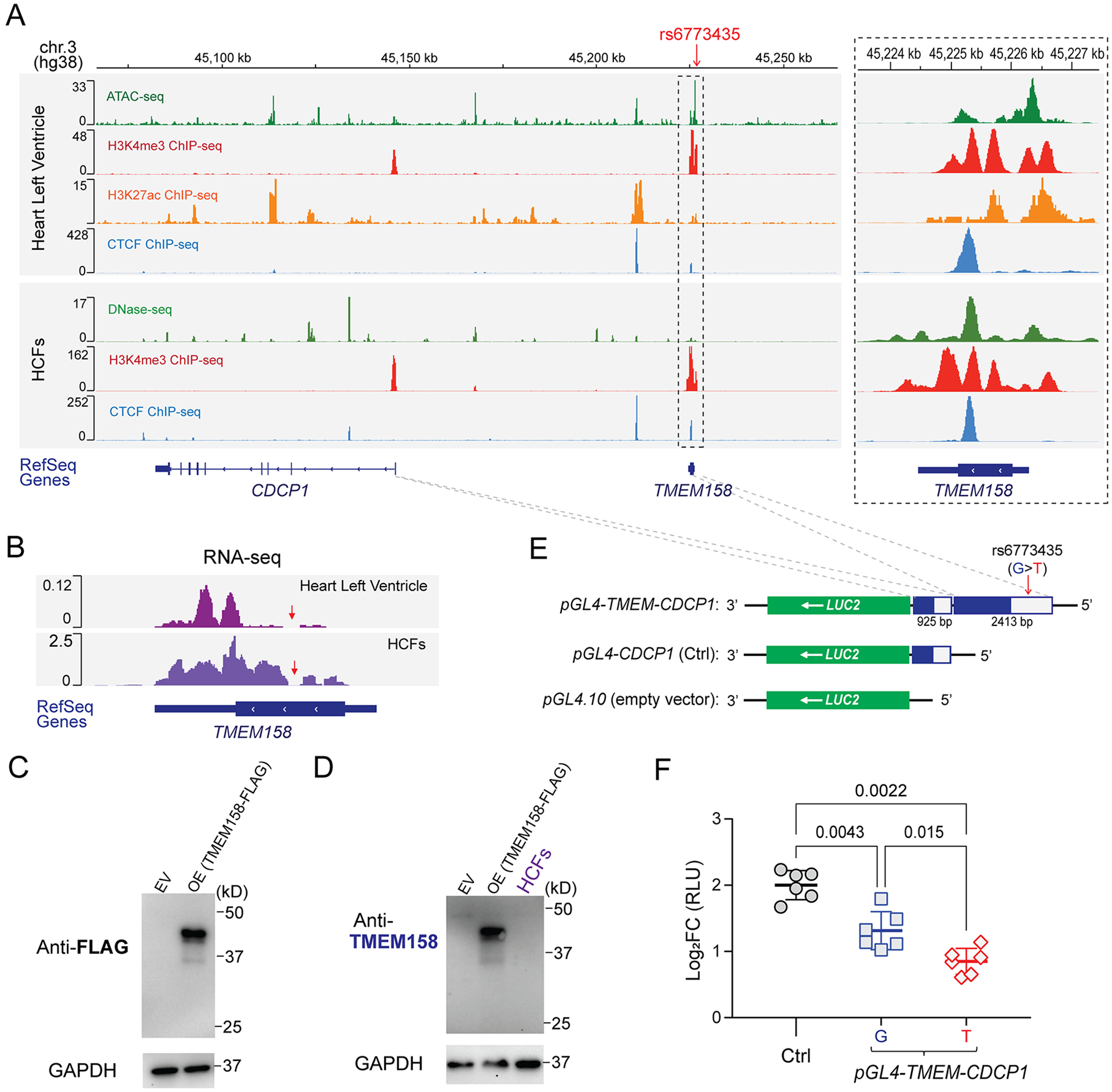
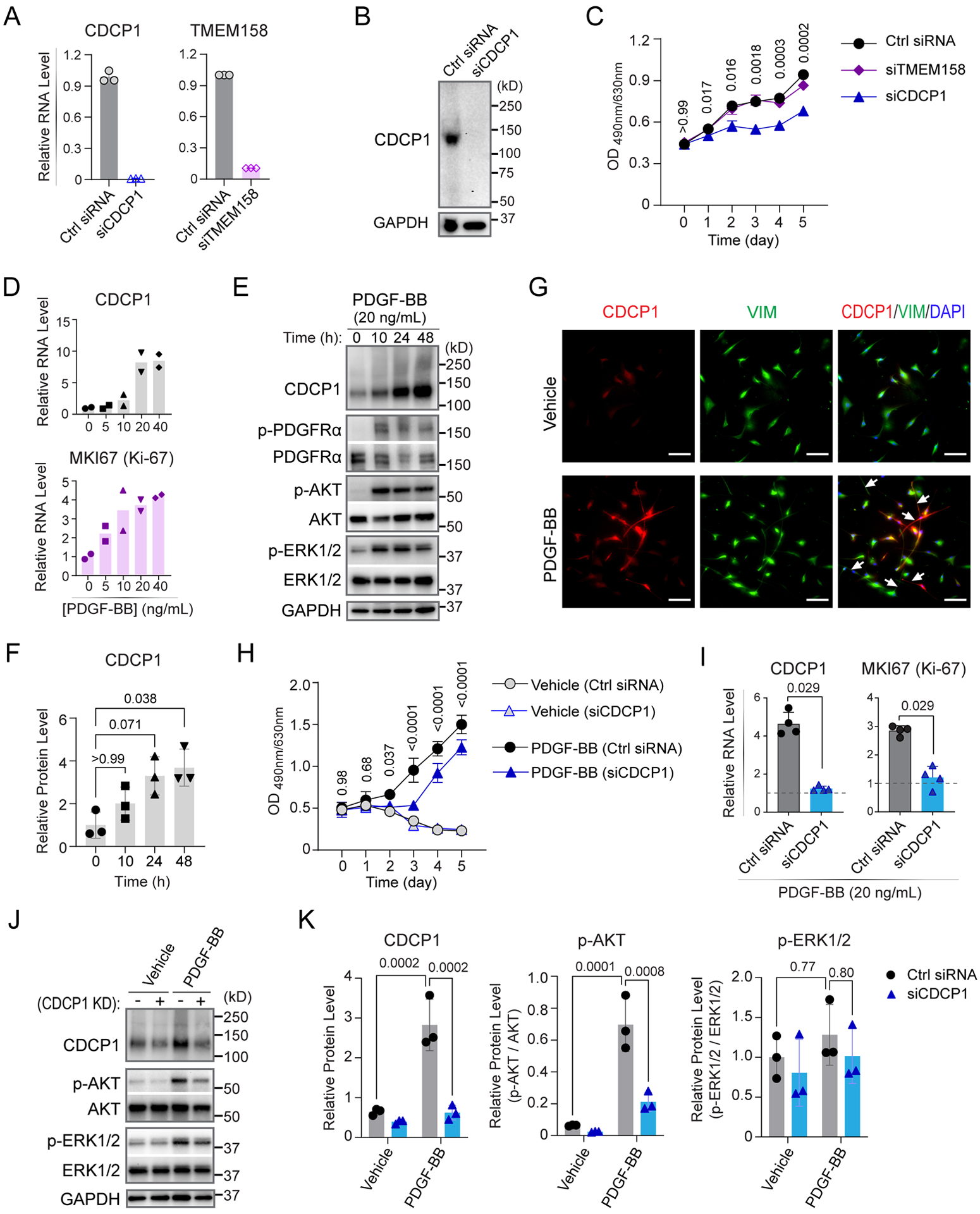
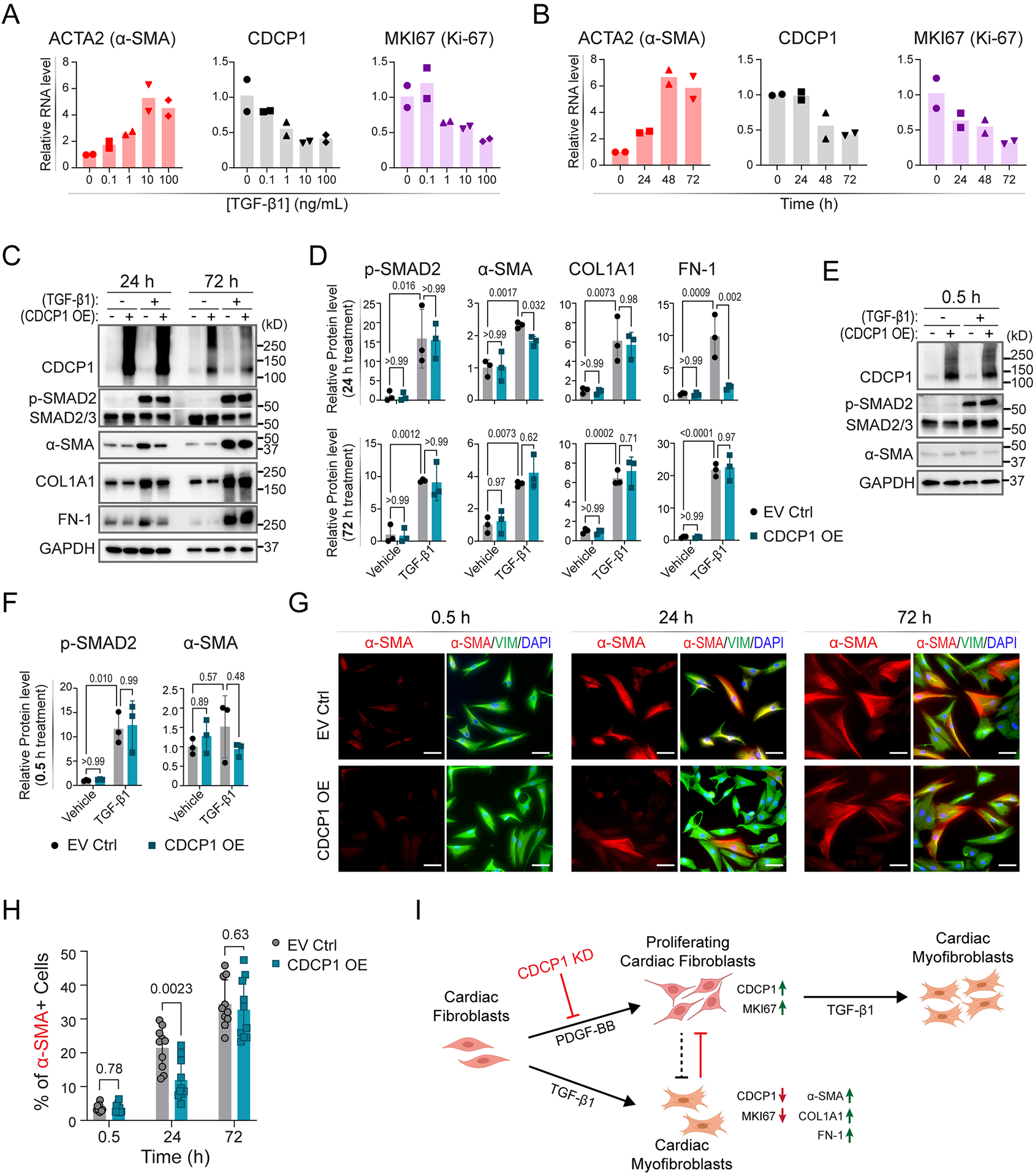
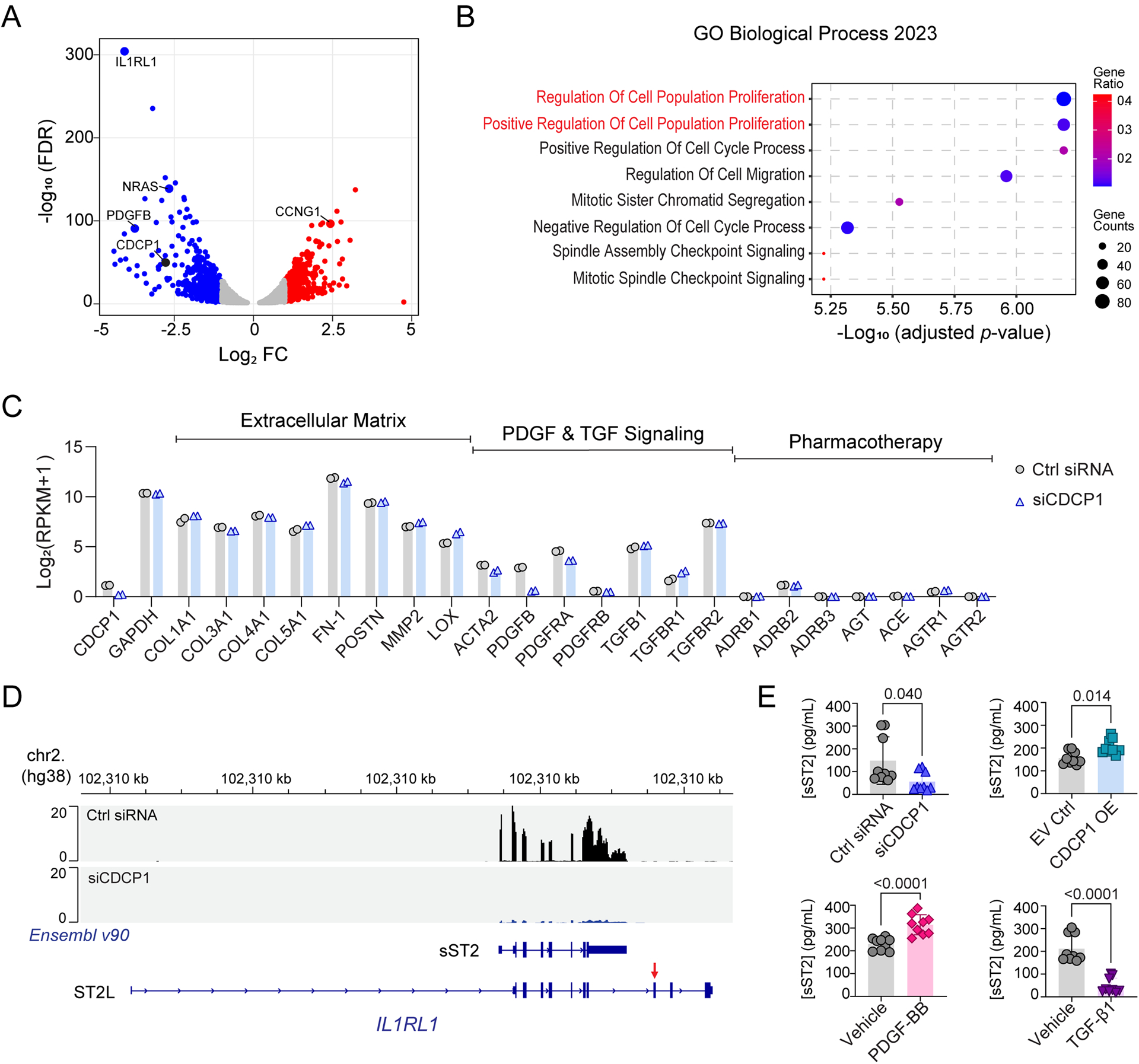
Results: The genome-wide association study identified a highly suggestive locus that mapped to the 5'-flanking region of the CDCP1 (CUB [complement C1r/C1s, Uegf, and Bmp1] domain containing protein 1) gene (rs6773435; P=7.12×10-7). The variant allele was associated with improved cardiac function and decreased CDCP1 transcription. CDCP1 expression was significantly upregulated in human cardiac fibroblasts (HCFs) in response to the PDGF (platelet-derived growth factor) signaling, and knockdown of CDCP1 significantly repressed HCF proliferation and decreased AKT (protein kinase B) phosphorylation. Transcriptomic profiling after CDCP1 knockdown in HCFs supported the conclusion that CDCP1 regulates HCF proliferation and mitosis. In addition, CDCP1 knockdown in HCFs resulted in significantly decreased expression of soluble ST2 (suppression of tumorigenicity-2), a prognostic biomarker for heart failure and inductor of cardiac fibrosis.
Conclusions: CDCP1 may play an important role in myocardial recovery in recent-onset DCM and mediates its effect primarily by attenuating cardiac fibrosis.
Keywords: cardiomyopathy, dilated; fibrosis; genetics; genome-wide association study; heart failure; humans; ventricular remodeling.
Conflict of interest statement
Figures
References
-
- McNamara DM, Starling RC, Cooper LT, Boehmer JP, Mather PJ, Janosko KM, Gorcsan J 3rd, Kip KE, Dec GW, Investigators I. Clinical and demographic predictors of outcomes in recent onset dilated cardiomyopathy: results of the IMAC (Intervention in Myocarditis and Acute Cardiomyopathy)-2 study. J Am Coll Cardiol. 2011;58:1112–1118. doi: 10.1016/j.jacc.2011.05.033 - DOI - PMC - PubMed
-
- Kramer DG, Trikalinos TA, Kent DM, Antonopoulos GV, Konstam MA, Udelson JE. Quantitative evaluation of drug or device effects on ventricular remodeling as predictors of therapeutic effects on mortality in patients with heart failure and reduced ejection fraction: a meta-analytic approach. J Am Coll Cardiol. 2010;56:392–406. doi: 10.1016/j.jacc.2010.05.011 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
