

Heterozygous deletion of the autism-associated gene CHD8 impairs synaptic function through widespread changes in gene expression and chromatin compaction
- PMID: 37802044
- PMCID: PMC10577079
- DOI: 10.1016/j.ajhg.2023.09.004
Heterozygous deletion of the autism-associated gene CHD8 impairs synaptic function through widespread changes in gene expression and chromatin compaction
Abstract
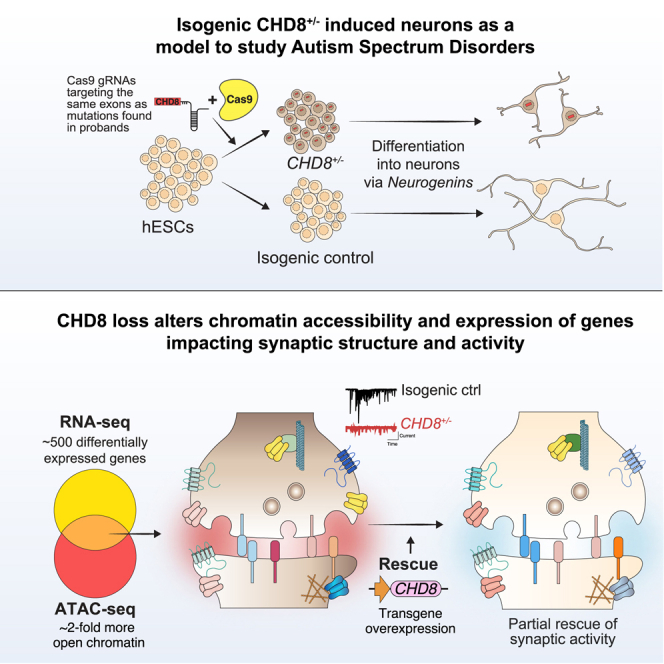
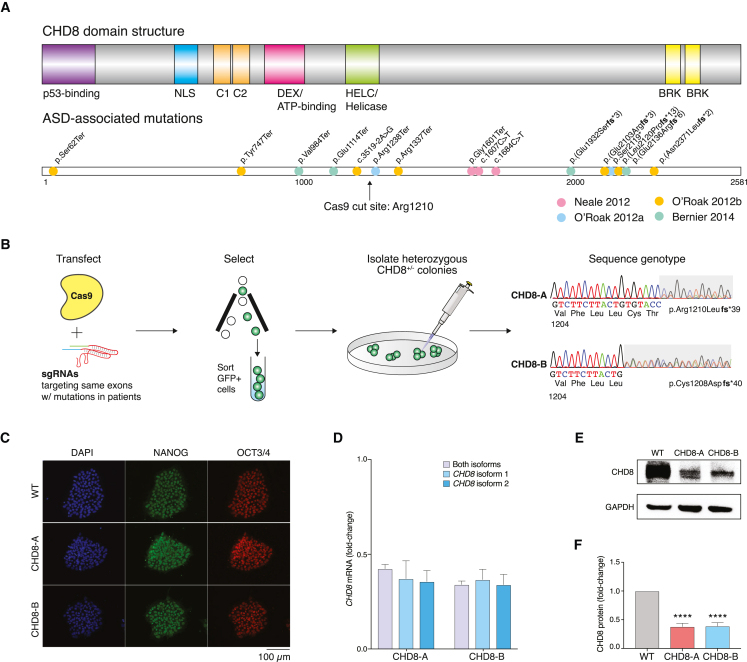
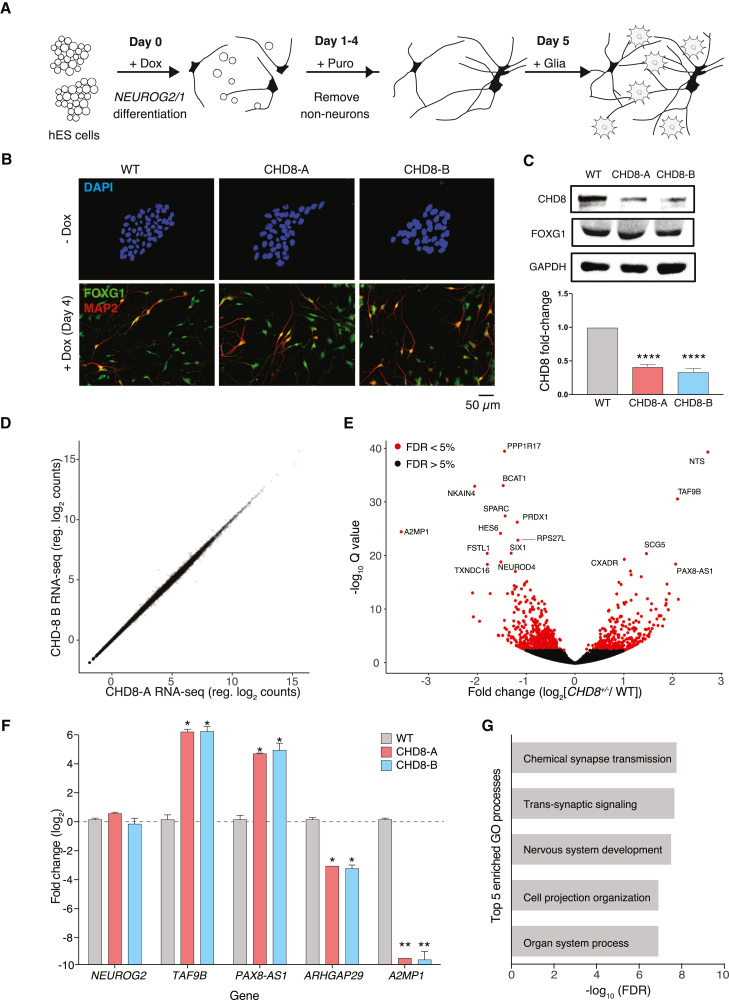
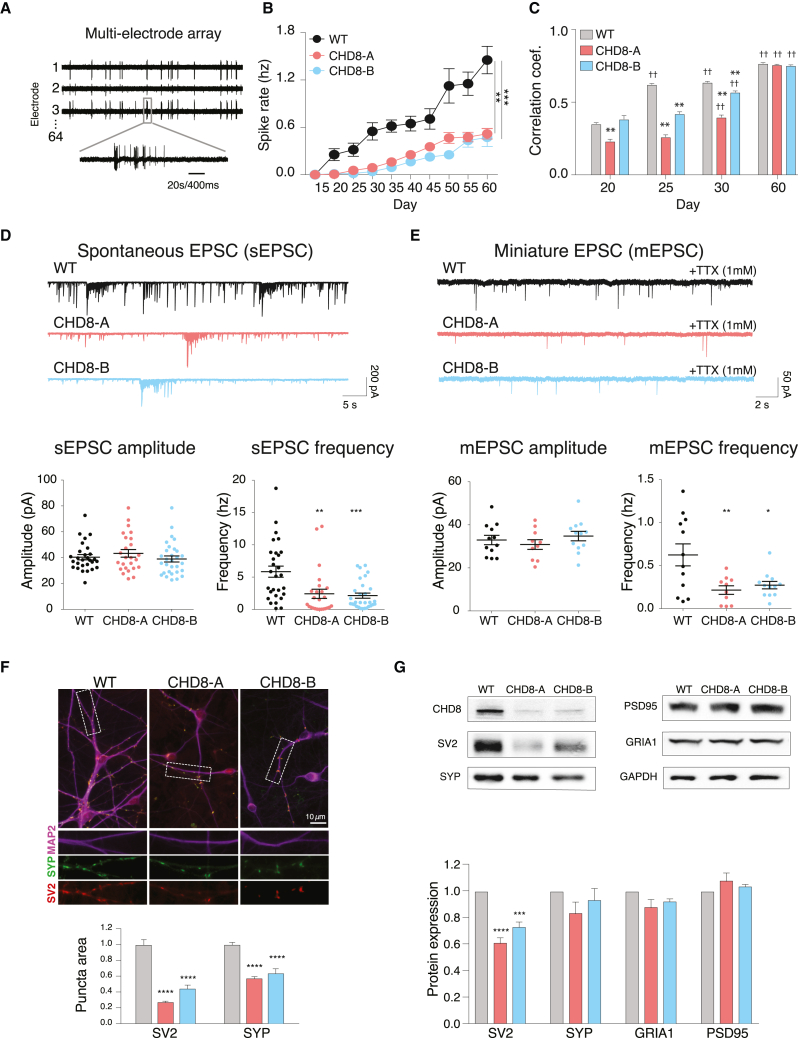
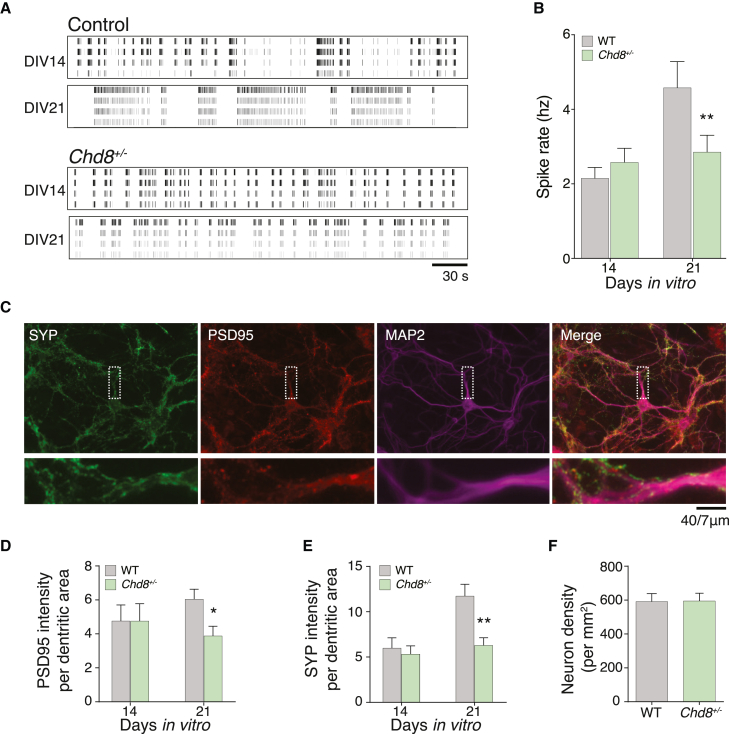
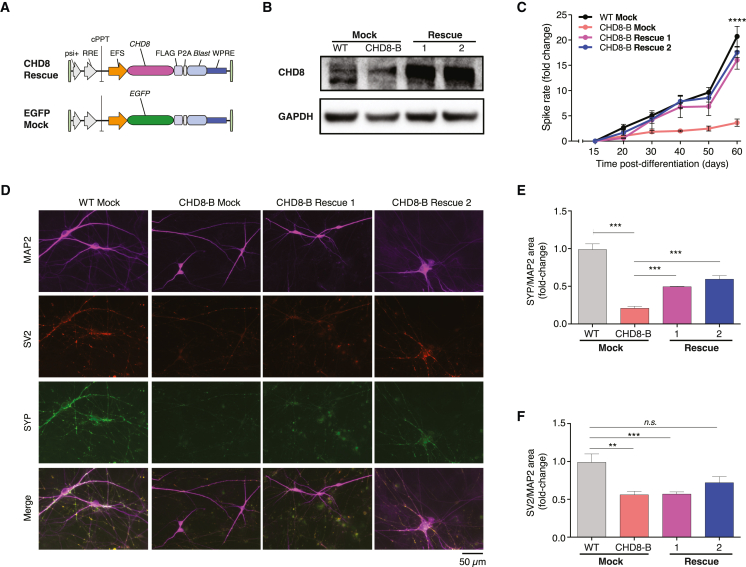
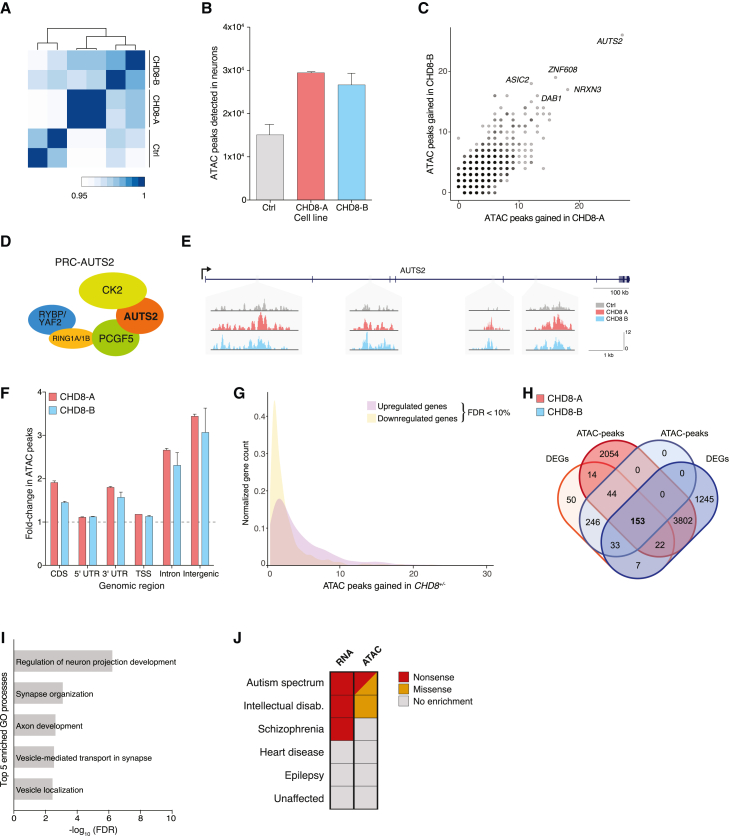
Whole-exome sequencing of autism spectrum disorder (ASD) probands and unaffected family members has identified many genes harboring de novo variants suspected to play a causal role in the disorder. Of these, chromodomain helicase DNA-binding protein 8 (CHD8) is the most recurrently mutated. Despite the prevalence of CHD8 mutations, we have little insight into how CHD8 loss affects genome organization or the functional consequences of these molecular alterations in neurons. Here, we engineered two isogenic human embryonic stem cell lines with CHD8 loss-of-function mutations and characterized differences in differentiated human cortical neurons. We identified hundreds of genes with altered expression, including many involved in neural development and excitatory synaptic transmission. Field recordings and single-cell electrophysiology revealed a 3-fold decrease in firing rates and synaptic activity in CHD8+/- neurons, as well as a similar firing-rate deficit in primary cortical neurons from Chd8+/- mice. These alterations in neuron and synapse function can be reversed by CHD8 overexpression. Moreover, CHD8+/- neurons displayed a large increase in open chromatin across the genome, where the greatest change in compaction was near autism susceptibility candidate 2 (AUTS2), which encodes a transcriptional regulator implicated in ASD. Genes with changes in chromatin accessibility and expression in CHD8+/- neurons have significant overlap with genes mutated in probands for ASD, intellectual disability, and schizophrenia but not with genes mutated in healthy controls or other disease cohorts. Overall, this study characterizes key molecular alterations in genome structure and expression in CHD8+/- neurons and links these changes to impaired neuronal and synaptic function.
Keywords: ASD; CHD8; CRISPR; autism; chromatin; chromodomain; helicase; isogenic; neurodevelopment; synaptic transmission.
Copyright © 2023 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.Z. is a scientific advisor to and cofounder of Editas Medicine, Beam Therapeutics, Pairwise Plants, Arbor Biotechnologies, Proof Diagnostics, and Aera Therapeutics. F.Z. is a scientific advisor to Octant. N.E.S. is a scientific advisor to Qiagen and is a scientific advisor to and cofounder of OverT Bio.
Figures
References
-
- Baio J., Wiggins L., Christensen D.L., Maenner M.J., Daniels J., Warren Z., Kurzius-Spencer M., Zahorodny W., Robinson Rosenberg C., White T., et al. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR. Surveill. Summ. 2018;67:1–23. doi: 10.15585/mmwr.ss6706a1. - DOI - PMC - PubMed
-
- Sanders S.J., He X., Willsey A.J., Ercan-Sencicek A.G., Samocha K.E., Cicek A.E., Murtha M.T., Bal V.H., Bishop S.L., Dong S., et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
