

A pharmacophore-guided deep learning approach for bioactive molecular generation
- PMID: 37803000
- PMCID: PMC10558534
- DOI: 10.1038/s41467-023-41454-9
A pharmacophore-guided deep learning approach for bioactive molecular generation
Abstract
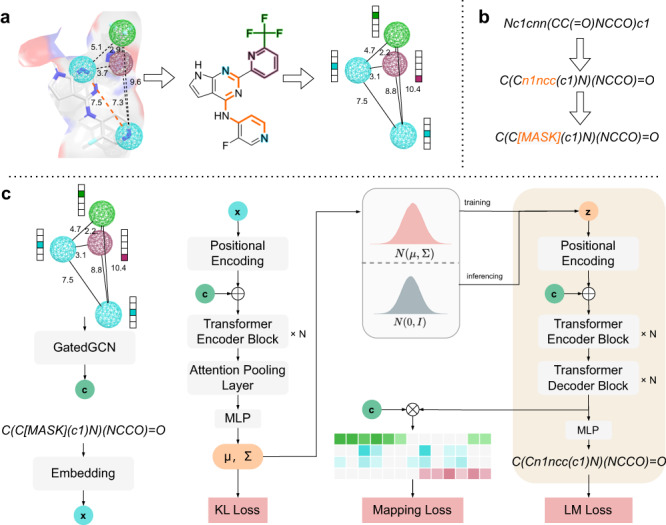
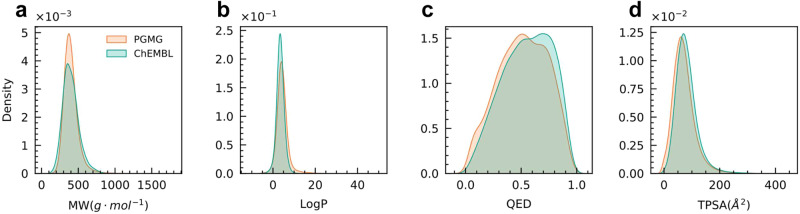
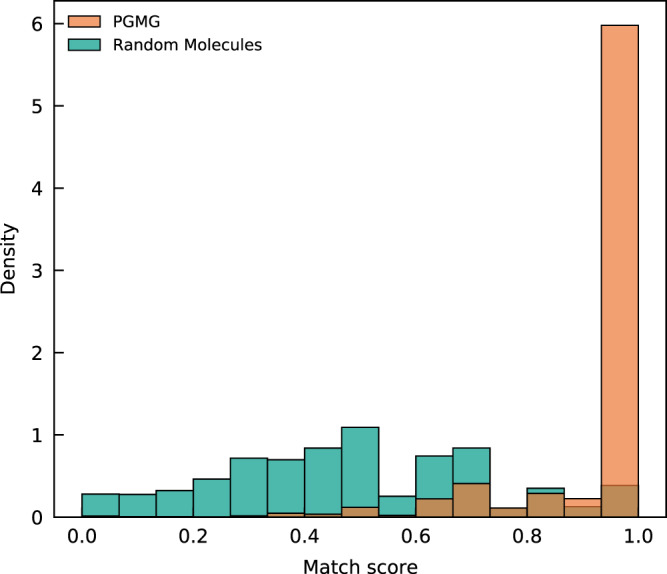
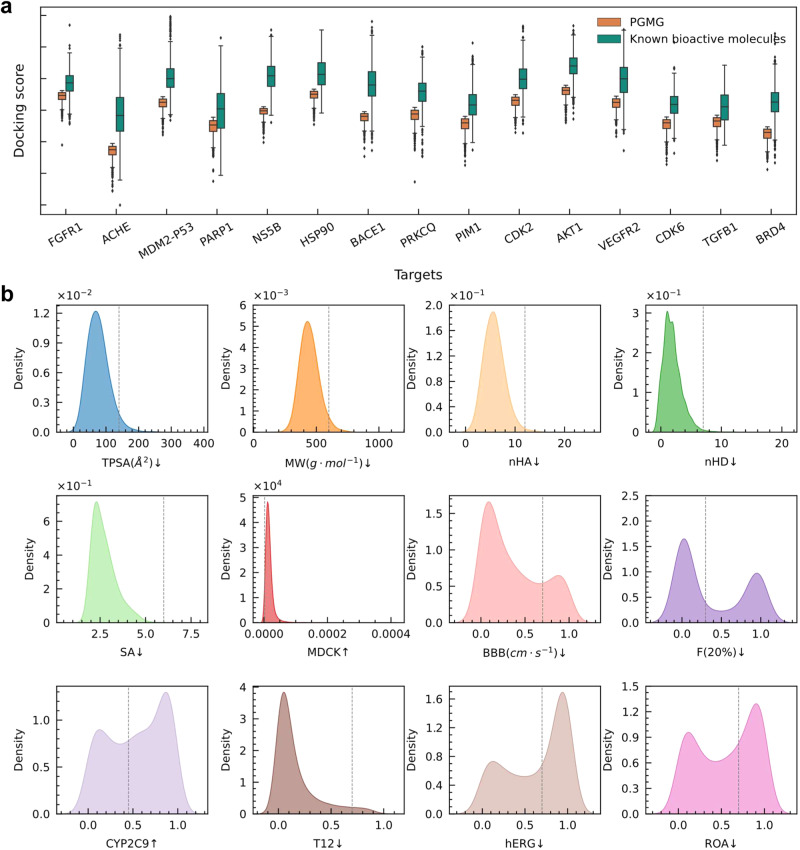
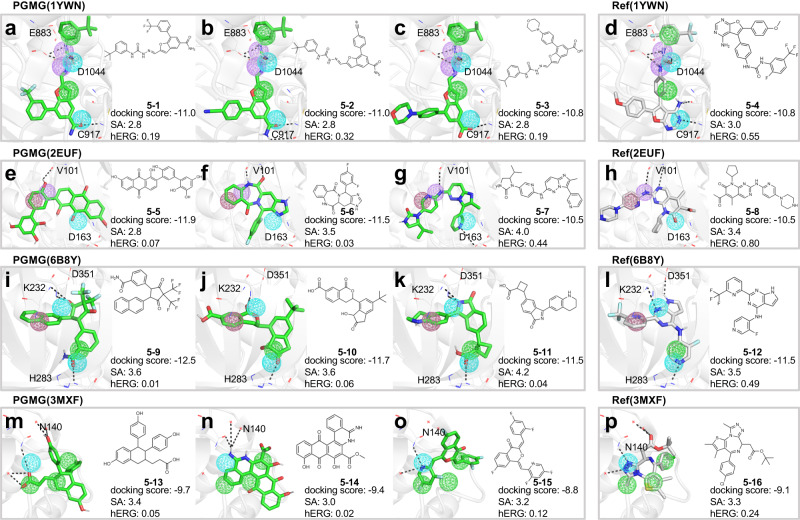
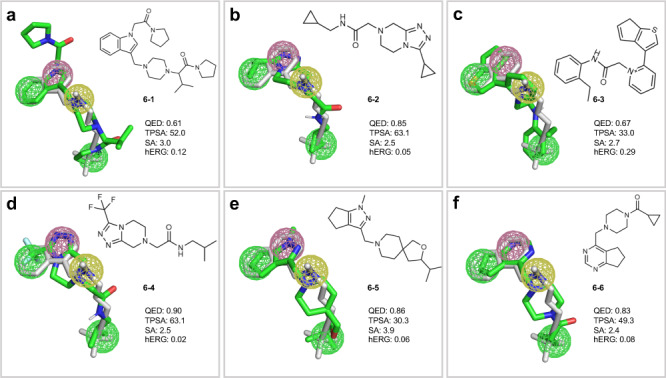
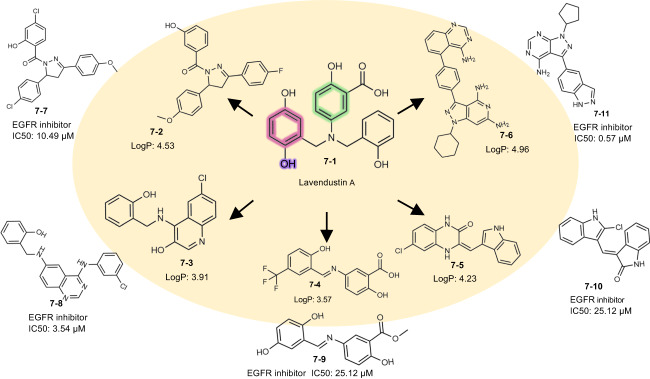
The rational design of novel molecules with the desired bioactivity is a critical but challenging task in drug discovery, especially when treating a novel target family or understudied targets. We propose a Pharmacophore-Guided deep learning approach for bioactive Molecule Generation (PGMG). Through the guidance of pharmacophore, PGMG provides a flexible strategy for generating bioactive molecules. PGMG uses a graph neural network to encode spatially distributed chemical features and a transformer decoder to generate molecules. A latent variable is introduced to solve the many-to-many mapping between pharmacophores and molecules to improve the diversity of the generated molecules. Compared to existing methods, PGMG generates molecules with strong docking affinities and high scores of validity, uniqueness, and novelty. In the case studies, we use PGMG in a ligand-based and structure-based drug de novo design. Overall, the flexibility and effectiveness make PGMG a useful tool to accelerate the drug discovery process.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. - PubMed
-
- Bohacek RS, McMartin C, Guida WC. The art and practice of structure‐based drug design: a molecular modeling perspective. Med. Res. Rev. 1996;16:3–50. - PubMed
-
- Goodnow RA., Jr Hit and lead identification: Integrated technology-based approaches. Drug Discov. Today. Technol. 2006;3:367–375.
-
- Jin W, Barzilay R, Jaakkola T. Multi-objective molecule generation using interpretable substructures. Int. Conf. Mach. Learn. 2020;37:4849–4859.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
