

Understanding and leveraging phenotypic plasticity during metastasis formation
- PMID: 37803056
- PMCID: PMC10558468
- DOI: 10.1038/s41540-023-00309-1
Understanding and leveraging phenotypic plasticity during metastasis formation
Abstract
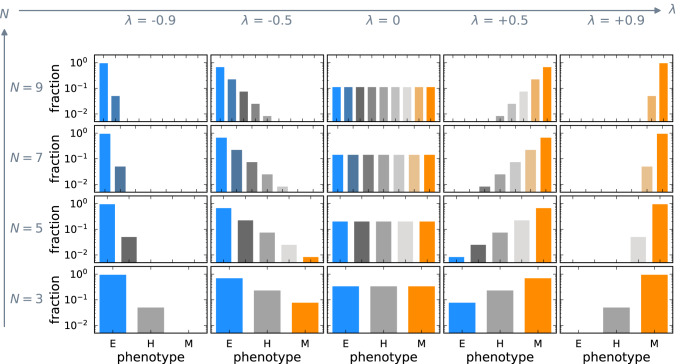
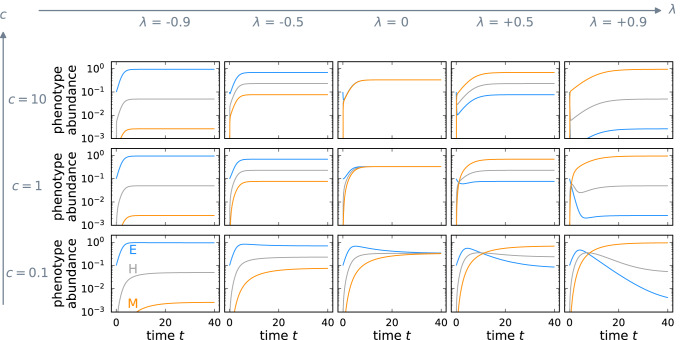
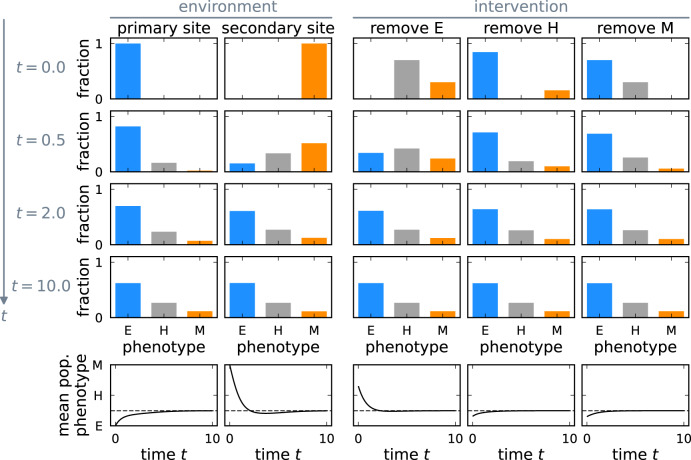
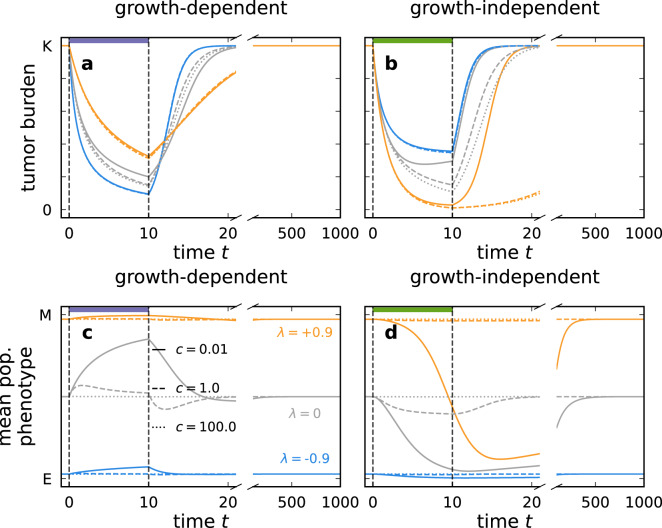
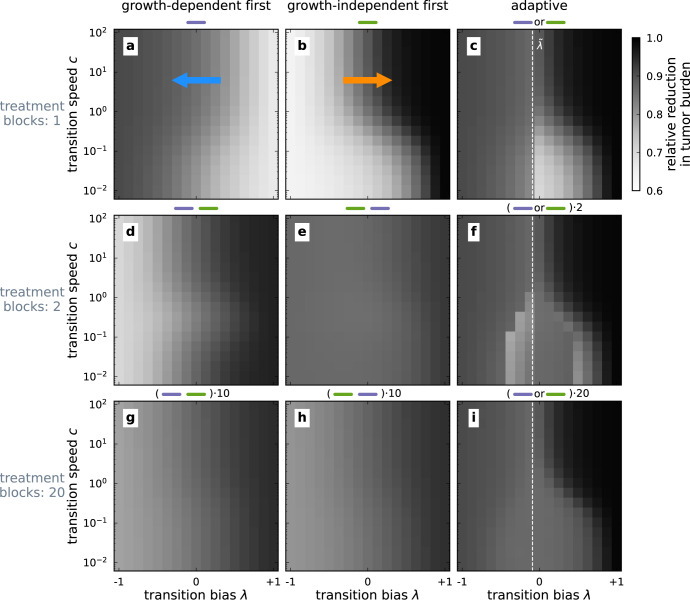
Cancer metastasis is the process of detrimental systemic spread and the primary cause of cancer-related fatalities. Successful metastasis formation requires tumor cells to be proliferative and invasive; however, cells cannot be effective at both tasks simultaneously. Tumor cells compensate for this trade-off by changing their phenotype during metastasis formation through phenotypic plasticity. Given the changing selection pressures and competitive interactions that tumor cells face, it is poorly understood how plasticity shapes the process of metastasis formation. Here, we develop an ecology-inspired mathematical model with phenotypic plasticity and resource competition between phenotypes to address this knowledge gap. We find that phenotypically plastic tumor cell populations attain a stable phenotype equilibrium that maintains tumor cell heterogeneity. Considering treatment types inspired by chemo- and immunotherapy, we highlight that plasticity can protect tumors against interventions. Turning this strength into a weakness, we corroborate current clinical practices to use plasticity as a target for adjuvant therapy. We present a parsimonious view of tumor plasticity-driven metastasis that is quantitative and experimentally testable, and thus potentially improving the mechanistic understanding of metastasis at the cell population level, and its treatment consequences.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. Cancer J. Clin. 2020;70:7–30. - PubMed
-
- Sung H, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Cancer J. Clin. 2021;71:209–249. - PubMed
-
- Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. - PubMed
-
- Weinberg, R. A. The Biology of Cancer 2nd edn (Garland Science, Taylor & Francis Group, 2014). ISBN 978-0-8153-4219-9 978-0-8153-4220-5.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
