

doi: 10.1021/acs.jcim.3c01153.
Epub 2023 Oct 8.
AmberTools
Affiliations
- PMID: 37805934
- PMCID: PMC10598796
- DOI: 10.1021/acs.jcim.3c01153
Item in Clipboard
AmberTools
J Chem Inf Model.
.
Abstract
AmberTools is a free and open-source collection of programs used to set up, run, and analyze molecular simulations. The newer features contained within AmberTools23 are briefly described in this Application note.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
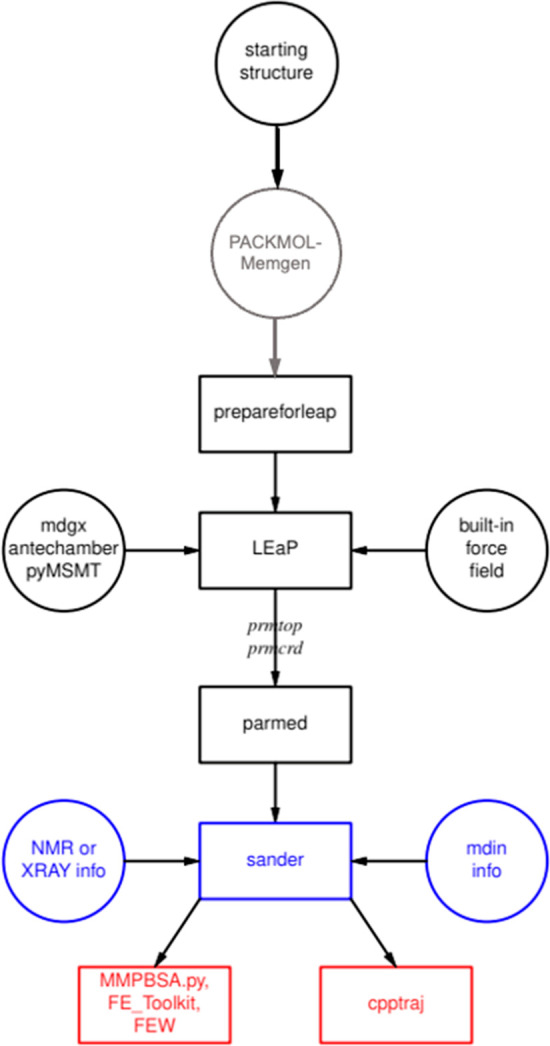
Common workflow in AmberTools. Flow went from top to bottom.
Black
boxes are for preparation, gray indicates an optional preparation
step specific for membrane systems, blue for simulation, and red for
analysis.
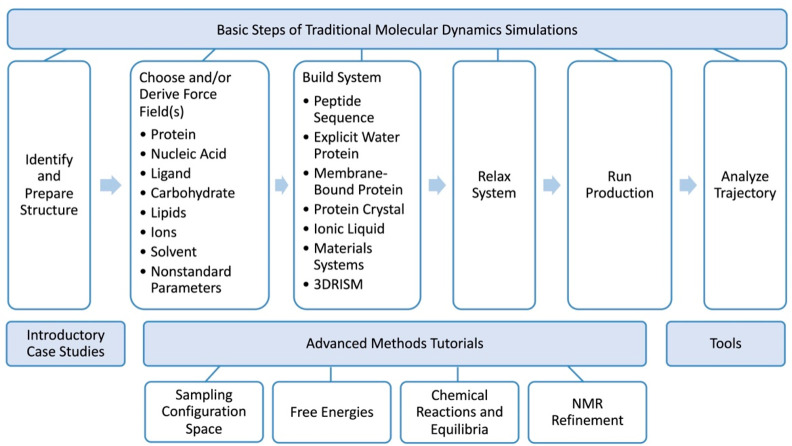
Overview of the Amber Tutorials. Tutorials are modular,
cover the basic steps of a typical molecular dynamics simulation,
introductory case studies, advanced methods, and some tools that are
commonly employed by Amber users.
References
-
- Weiner P. K.; Kollman P. A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. 10.1002/jcc.540020311. - DOI
-
- Pearlman D.; Case D. A.; Caldwell J.; Ross W. S.; Cheatham T. E. II; DeBolt S.; Ferguson D.; Seibel G.; Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. 10.1016/0010-4655(95)00041-D. - DOI
-
- Salomon-Ferrer R.; Case D. A.; Walker R. C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. 10.1002/wcms.1121. - DOI
Publication types
Grants and funding
- R35 GM144089/GM/NIGMS NIH HHS/United States
- R01 GM107485/GM/NIGMS NIH HHS/United States
- R01 GM107104/GM/NIGMS NIH HHS/United States
- R01 GM062248/GM/NIGMS NIH HHS/United States
- R01 GM108583/GM/NIGMS NIH HHS/United States
- R35 GM151951/GM/NIGMS NIH HHS/United States
- Z01 HL001051/ImNIH/Intramural NIH HHS/United States
- R01 GM149874/GM/NIGMS NIH HHS/United States
- R35 GM130367/GM/NIGMS NIH HHS/United States
- R01 GM144596/GM/NIGMS NIH HHS/United States
- R01 GM130641/GM/NIGMS NIH HHS/United States
- R01 GM147673/GM/NIGMS NIH HHS/United States
- RM1 GM135136/GM/NIGMS NIH HHS/United States
- R16 GM146633/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
