

Evaluation of whole and core genome multilocus sequence typing allele schemes for Salmonella enterica outbreak detection in a national surveillance network, PulseNet USA
- PMID: 37808298
- PMCID: PMC10558246
- DOI: 10.3389/fmicb.2023.1254777
Evaluation of whole and core genome multilocus sequence typing allele schemes for Salmonella enterica outbreak detection in a national surveillance network, PulseNet USA
Abstract
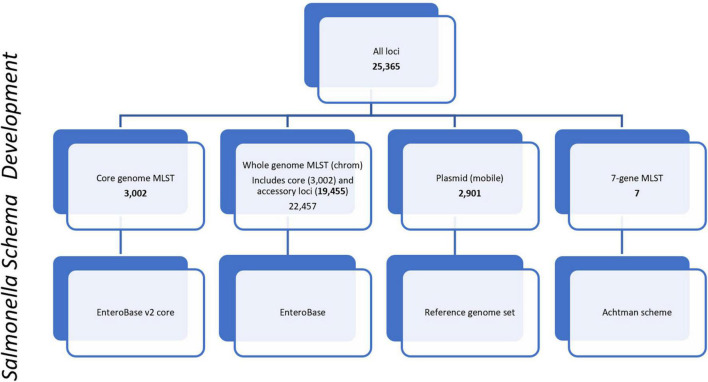
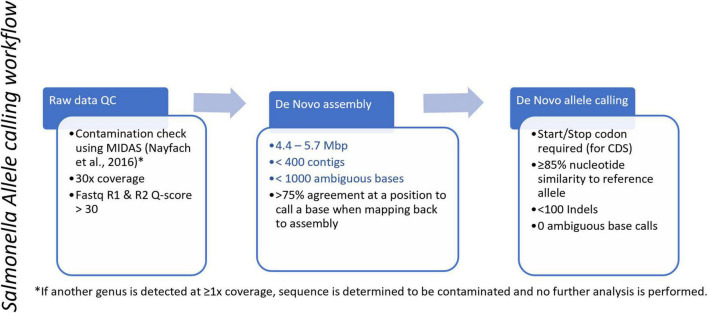
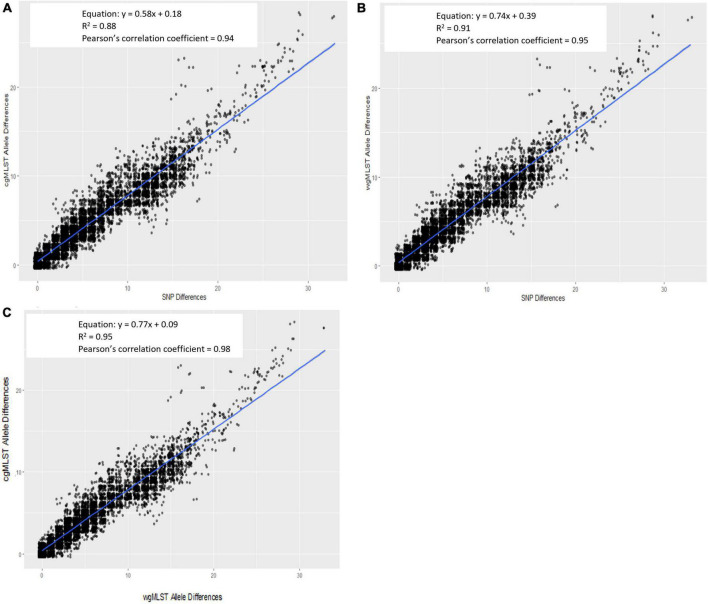
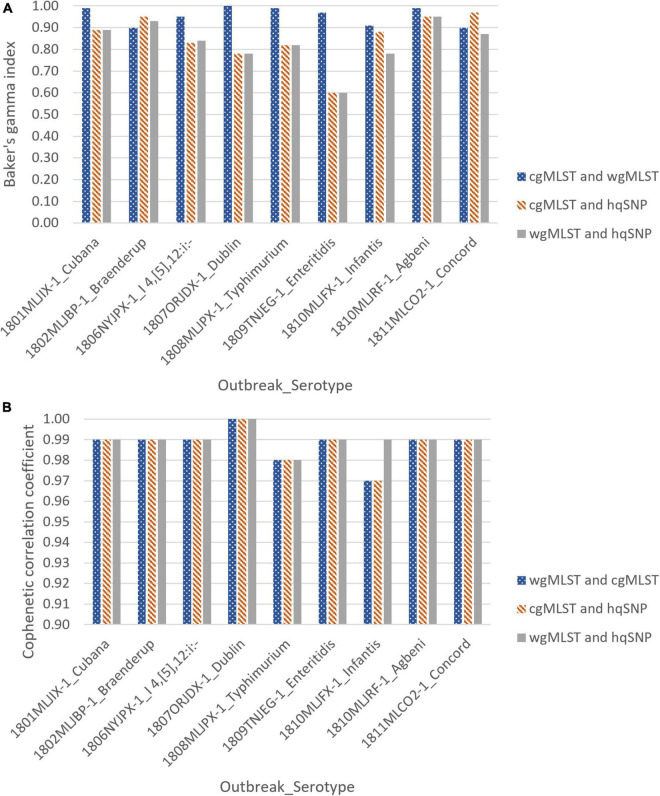
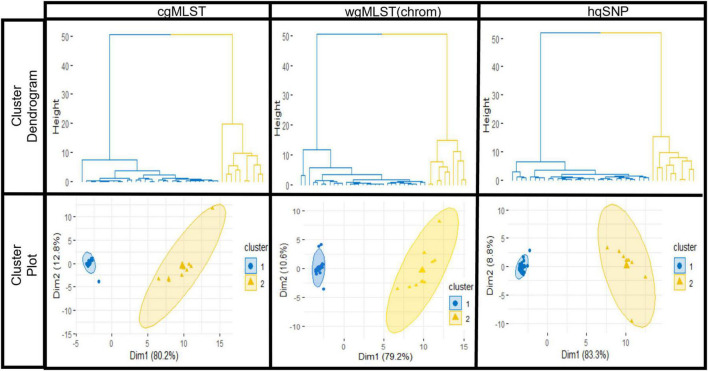
Salmonella enterica is a leading cause of bacterial foodborne and zoonotic illnesses in the United States. For this study, we applied four different whole genome sequencing (WGS)-based subtyping methods: high quality single-nucleotide polymorphism (hqSNP) analysis, whole genome multilocus sequence typing using either all loci [wgMLST (all loci)] and only chromosome-associated loci [wgMLST (chrom)], and core genome multilocus sequence typing (cgMLST) to a dataset of isolate sequences from 9 well-characterized Salmonella outbreaks. For each outbreak, we evaluated the genomic and epidemiologic concordance between hqSNP and allele-based methods. We first compared pairwise genomic differences using all four methods. We observed discrepancies in allele difference ranges when using wgMLST (all loci), likely caused by inflated genetic variation due to loci found on plasmids and/or other mobile genetic elements in the accessory genome. Therefore, we excluded wgMLST (all loci) results from any further comparisons in the study. Then, we created linear regression models and phylogenetic tanglegrams using the remaining three methods. K-means analysis using the silhouette method was applied to compare the ability of the three methods to partition outbreak and sporadic isolate sequences. Our results showed that pairwise hqSNP differences had high concordance with cgMLST and wgMLST (chrom) allele differences. The slopes of the regressions for hqSNP vs. allele pairwise differences were 0.58 (cgMLST) and 0.74 [wgMLST (chrom)], and the slope of the regression was 0.77 for cgMLST vs. wgMLST (chrom) pairwise differences. Tanglegrams showed high clustering concordance between methods using two statistical measures, the Baker's gamma index (BGI) and cophenetic correlation coefficient (CCC), where 9/9 (100%) of outbreaks yielded BGI values ≥ 0.60 and CCCs were ≥ 0.97 across all nine outbreaks and all three methods. K-means analysis showed separation of outbreak and sporadic isolate groups with average silhouette widths ≥ 0.87 for outbreak groups and ≥ 0.16 for sporadic groups. This study demonstrates that Salmonella isolates clustered in concordance with epidemiologic data using three WGS-based subtyping methods and supports using cgMLST as the primary method for national surveillance of Salmonella outbreak clusters.
Keywords: Salmonella; cgMLST; epidemiology; hqSNP; silhouette method; surveillance; wgMLST.
Copyright © 2023 Leeper, Tolar, Griswold, Vidyaprakash, Hise, Williams, Im, Chen, Pouseele and Carleton.
Conflict of interest statement
HP was employed by BioMérieux. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
References
-
- Baker F. (1974). Stability of two hierarchical grouping techniques case 1: Sensitivity to data errors. J. Am. Stat. Assoc. 69 440–445.
LinkOut - more resources
Full Text Sources