

This is a preprint.
Digital profiling of cancer transcriptomes from histology images with grouped vision attention
- PMID: 37808782
- PMCID: PMC10557714
- DOI: 10.1101/2023.09.28.560068
Digital profiling of cancer transcriptomes from histology images with grouped vision attention
Update in
-
Digital profiling of gene expression from histology images with linearized attention.Nat Commun. 2024 Nov 14;15(1):9886. doi: 10.1038/s41467-024-54182-5. Nat Commun. 2024. PMID: 39543087 Free PMC article.
Abstract
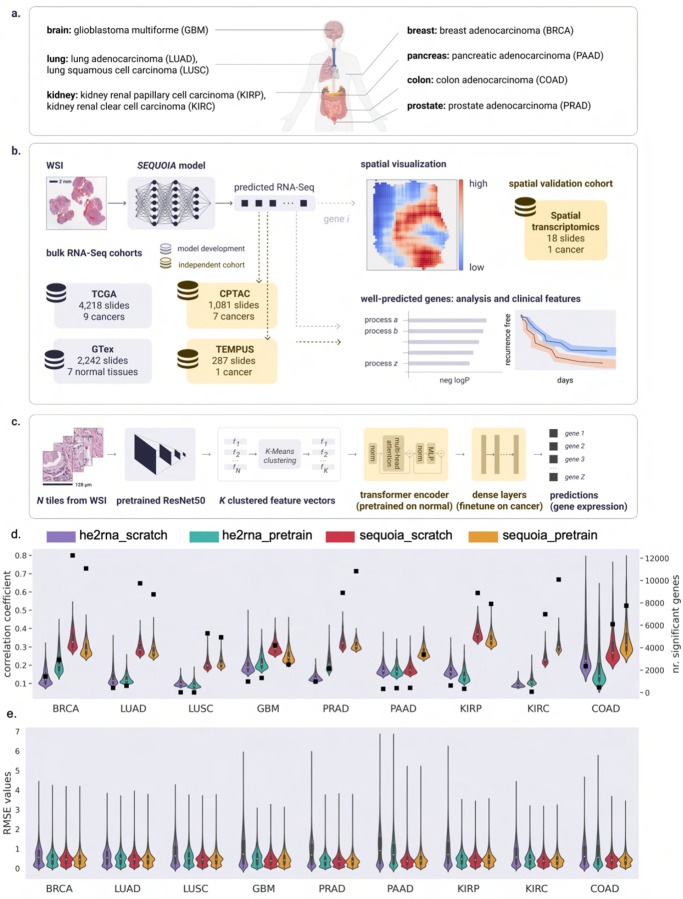
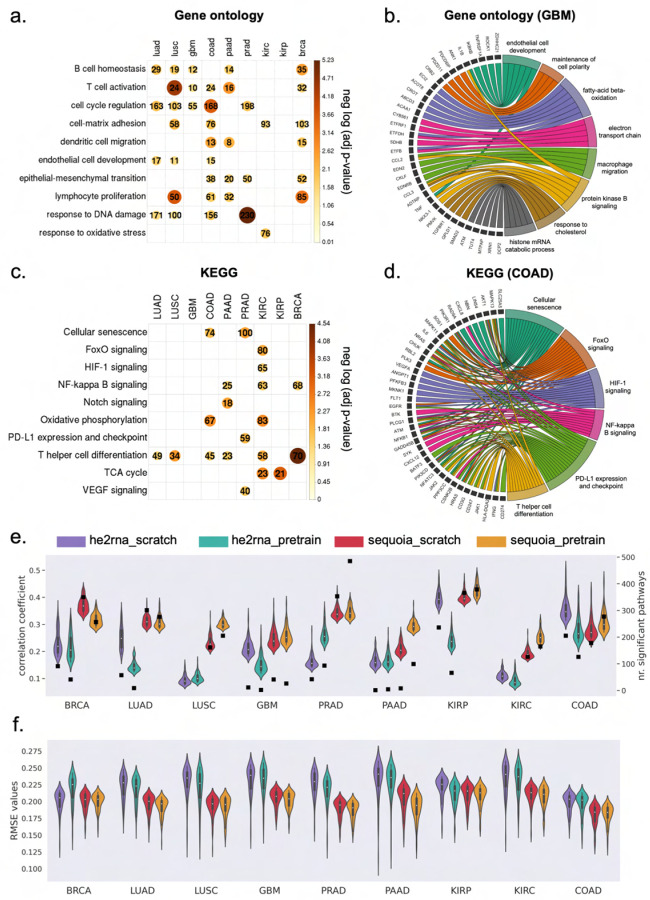
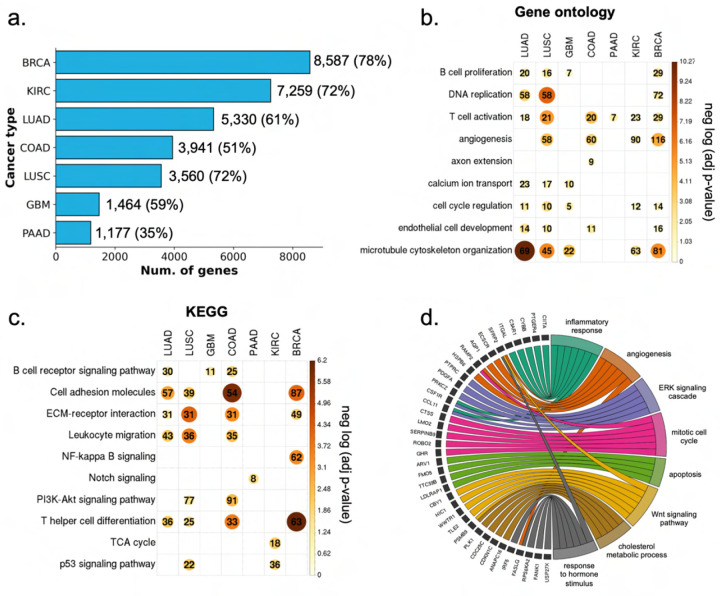
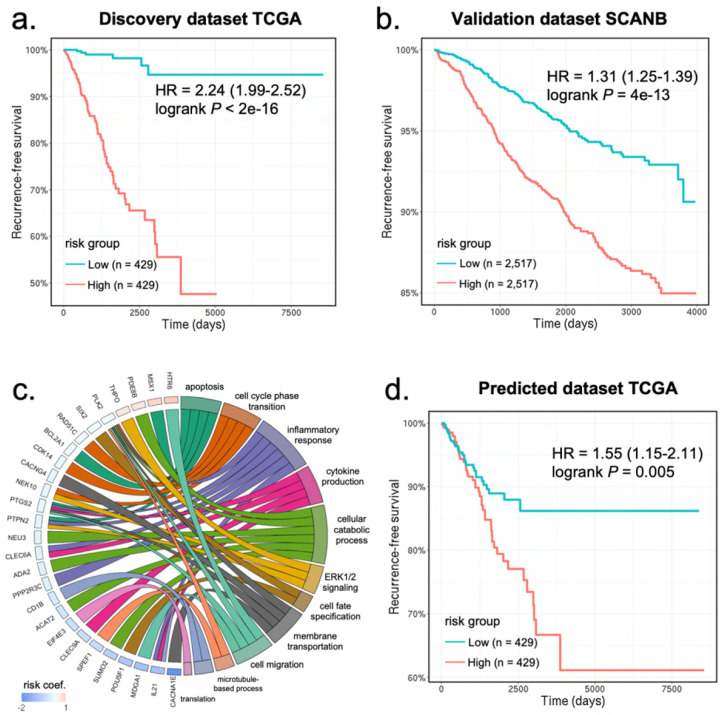
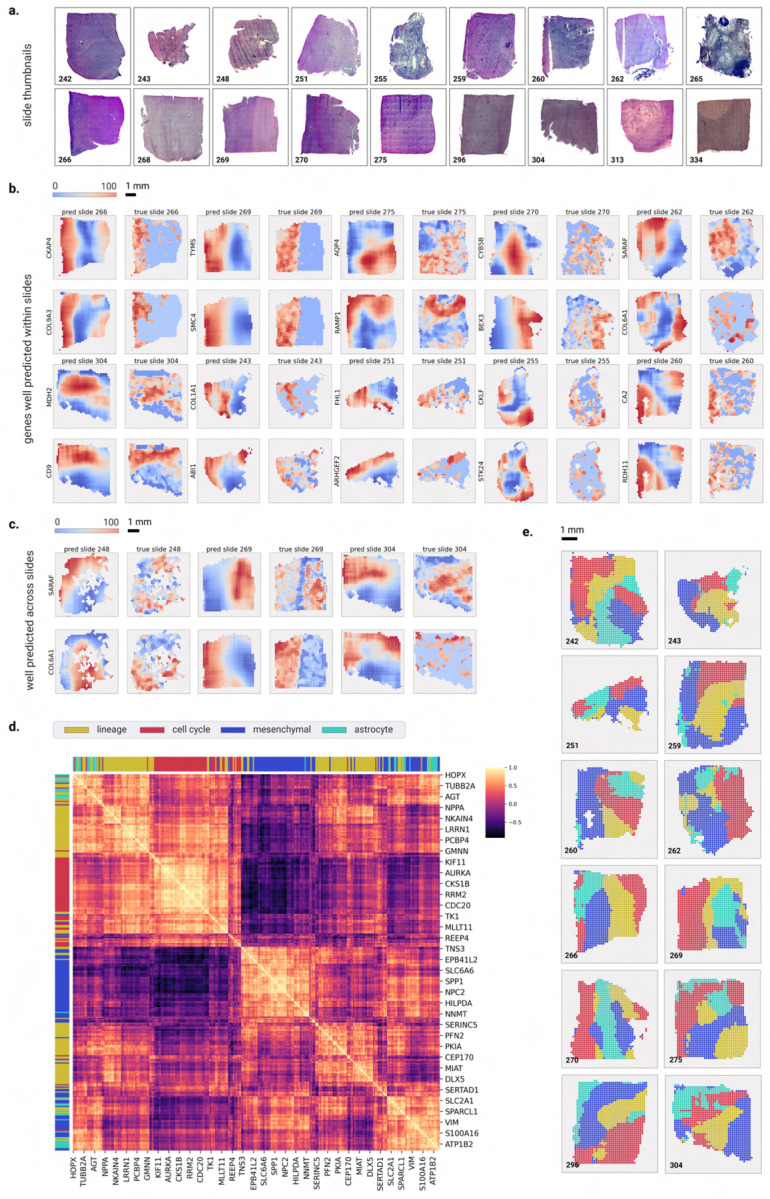
Cancer is a heterogeneous disease that demands precise molecular profiling for better understanding and management. Recently, deep learning has demonstrated potentials for cost-efficient prediction of molecular alterations from histology images. While transformer-based deep learning architectures have enabled significant progress in non-medical domains, their application to histology images remains limited due to small dataset sizes coupled with the explosion of trainable parameters. Here, we develop SEQUOIA, a transformer model to predict cancer transcriptomes from whole-slide histology images. To enable the full potential of transformers, we first pre-train the model using data from 1,802 normal tissues. Then, we fine-tune and evaluate the model in 4,331 tumor samples across nine cancer types. The prediction performance is assessed at individual gene levels and pathway levels through Pearson correlation analysis and root mean square error. The generalization capacity is validated across two independent cohorts comprising 1,305 tumors. In predicting the expression levels of 25,749 genes, the highest performance is observed in cancers from breast, kidney and lung, where SEQUOIA accurately predicts the expression of 11,069, 10,086 and 8,759 genes, respectively. The accurately predicted genes are associated with the regulation of inflammatory response, cell cycles and metabolisms. While the model is trained at the tissue level, we showcase its potential in predicting spatial gene expression patterns using spatial transcriptomics datasets. Leveraging the prediction performance, we develop a digital gene expression signature that predicts the risk of recurrence in breast cancer. SEQUOIA deciphers clinically relevant gene expression patterns from histology images, opening avenues for improved cancer management and personalized therapies.
Figures
References
-
- Hausser J., Alon U.: Tumour heterogeneity and the evolutionary trade-offs of cancer. Nature Reviews Cancer 20(4), 247–257 (2020) - PubMed
-
- Ravi V.M., Will P., Kueckelhaus J., Sun N., Joseph K., Salié H., Vollmer L., Kuliesiute U., Ehr J., Benotmane J.K., et al.: Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 40(6), 639–655 (2022) - PubMed
Publication types
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources