

Subclonal Somatic Copy-Number Alterations Emerge and Dominate in Recurrent Osteosarcoma
- PMID: 37812025
- PMCID: PMC10646480
- DOI: 10.1158/0008-5472.CAN-23-0385
Subclonal Somatic Copy-Number Alterations Emerge and Dominate in Recurrent Osteosarcoma
Abstract
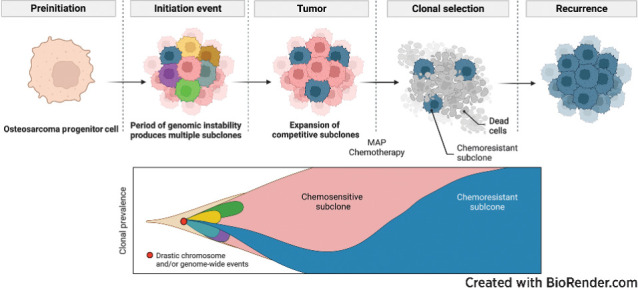
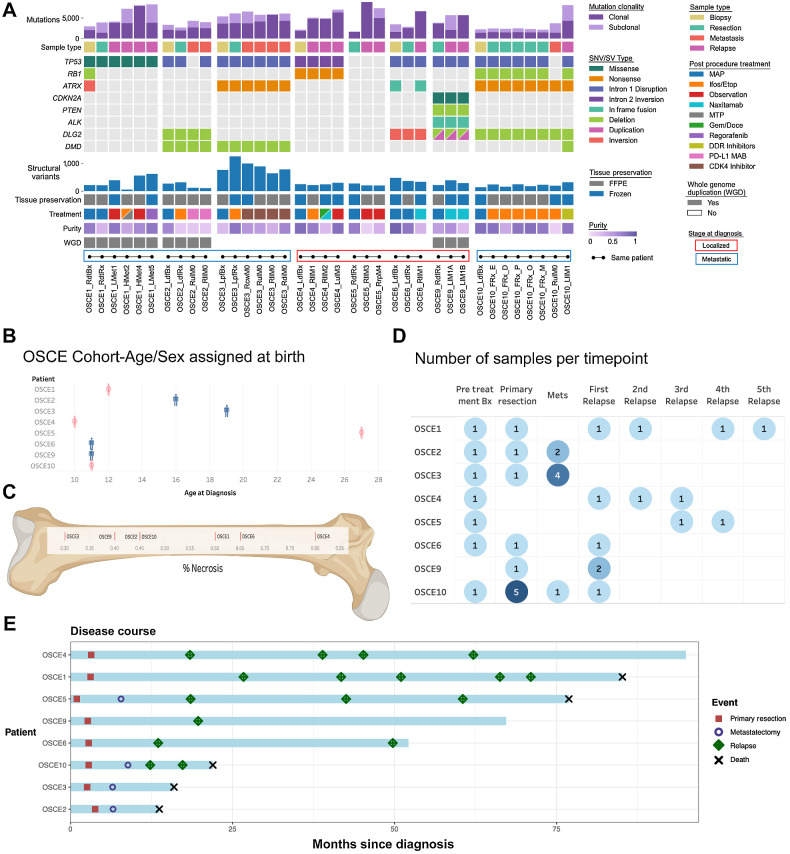
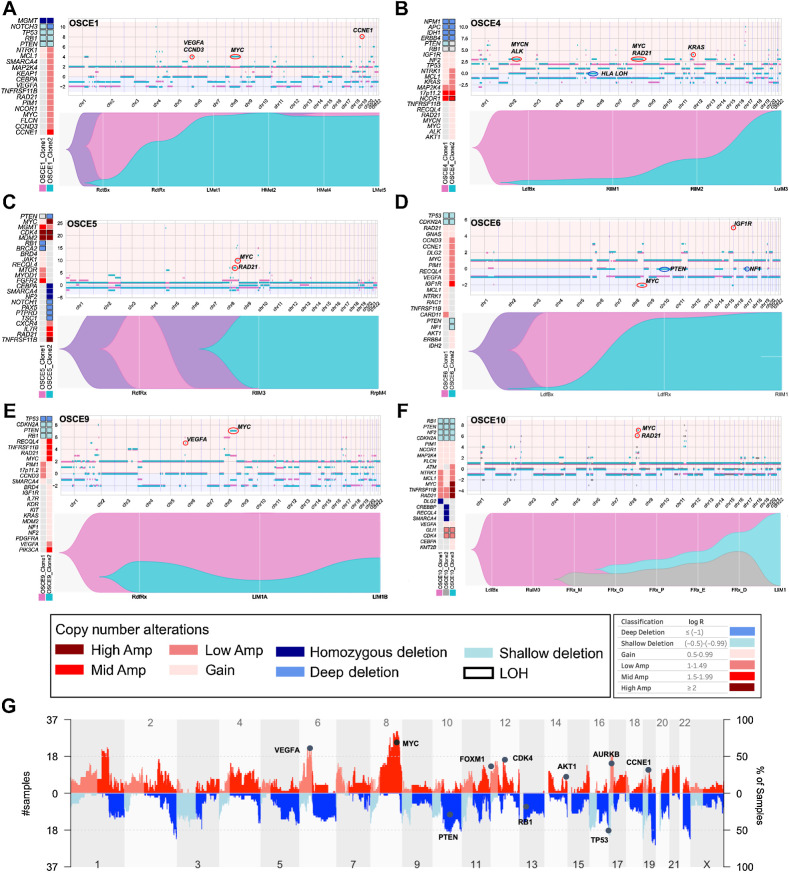
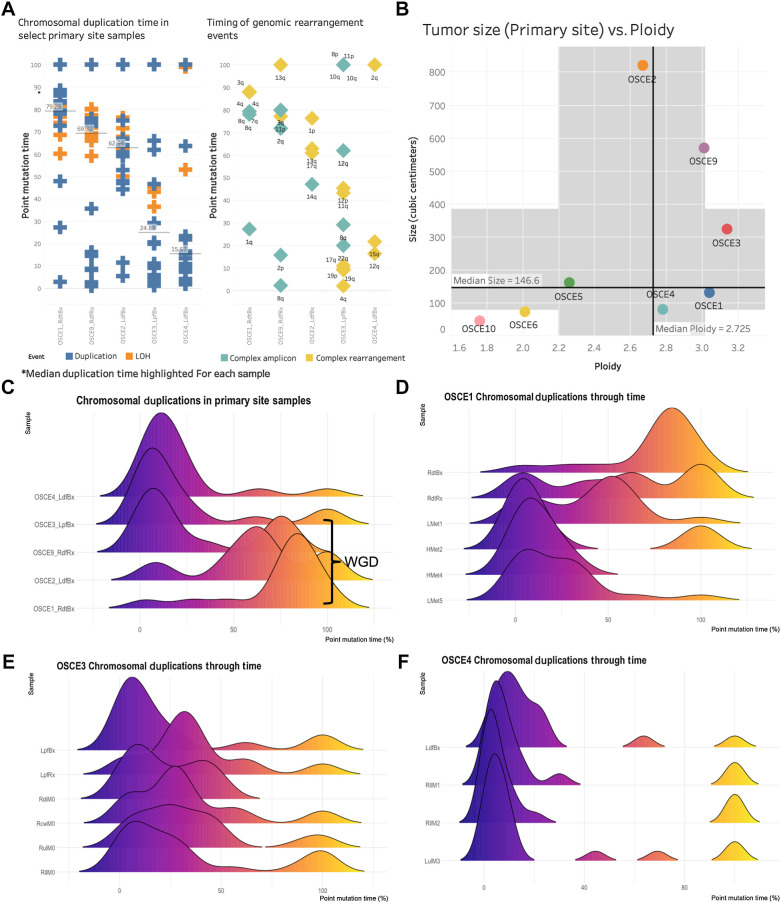
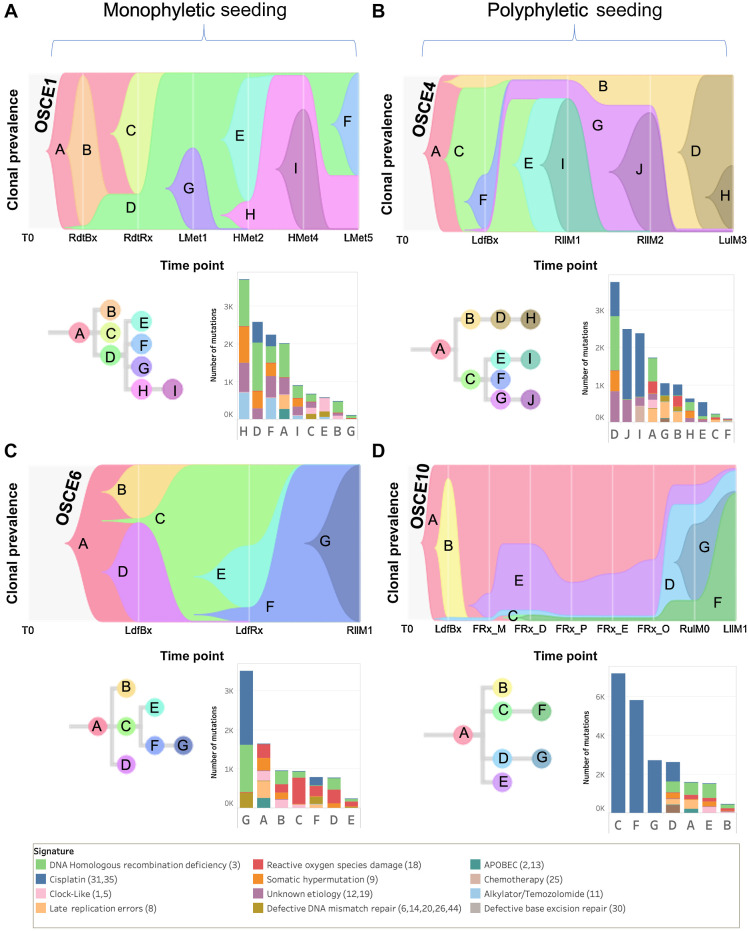
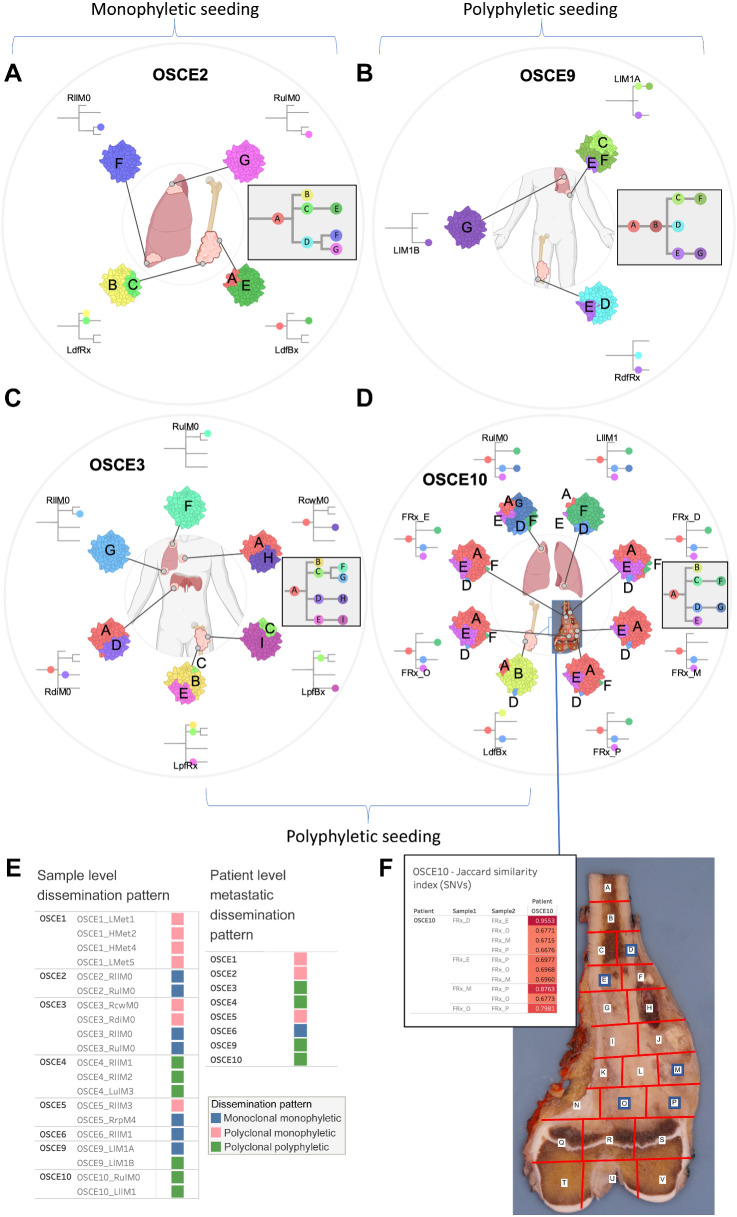
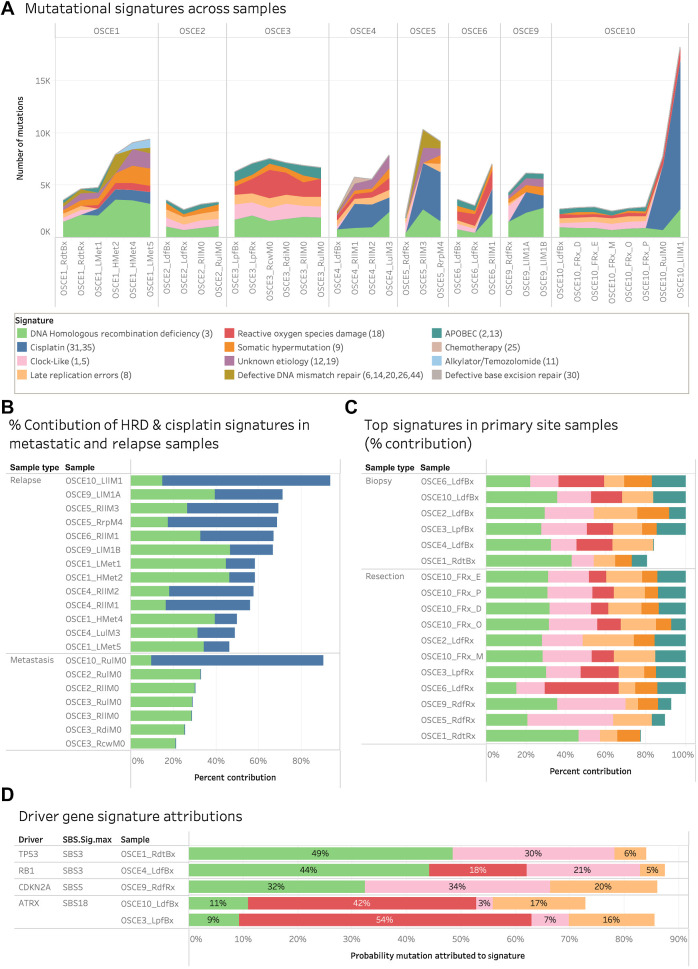
Multiple large-scale genomic profiling efforts have been undertaken in osteosarcoma to define the genomic drivers of tumorigenesis, therapeutic response, and disease recurrence. The spatial and temporal intratumor heterogeneity could also play a role in promoting tumor growth and treatment resistance. We conducted longitudinal whole-genome sequencing of 37 tumor samples from 8 patients with relapsed or refractory osteosarcoma. Each patient had at least one sample from a primary site and a metastatic or relapse site. Subclonal copy-number alterations were identified in all patients except one. In 5 patients, subclones from the primary tumor emerged and dominated at subsequent relapses. MYC gain/amplification was enriched in the treatment-resistant clones in 6 of 7 patients with multiple clones. Amplifications in other potential driver genes, such as CCNE1, RAD21, VEGFA, and IGF1R, were also observed in the resistant copy-number clones. A chromosomal duplication timing analysis revealed that complex genomic rearrangements typically occurred prior to diagnosis, supporting a macroevolutionary model of evolution, where a large number of genomic aberrations are acquired over a short period of time followed by clonal selection, as opposed to ongoing evolution. A mutational signature analysis of recurrent tumors revealed that homologous repair deficiency (HRD)-related SBS3 increases at each time point in patients with recurrent disease, suggesting that HRD continues to be an active mutagenic process after diagnosis. Overall, by examining the clonal relationships between temporally and spatially separated samples from patients with relapsed/refractory osteosarcoma, this study sheds light on the intratumor heterogeneity and potential drivers of treatment resistance in this disease.
Significance: The chemoresistant population in recurrent osteosarcoma is subclonal at diagnosis, emerges at the time of primary resection due to selective pressure from neoadjuvant chemotherapy, and is characterized by unique oncogenic amplifications.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
Update of
-
Subclonal somatic copy number alterations emerge and dominate in recurrent osteosarcoma.bioRxiv [Preprint]. 2023 Jan 24:2023.01.05.522765. doi: 10.1101/2023.01.05.522765. bioRxiv. 2023. Update in: Cancer Res. 2023 Nov 15;83(22):3796-3812. doi: 10.1158/0008-5472.CAN-23-0385. PMID: 36711976 Free PMC article. Updated. Preprint.
References
-
- Kager L, Zoubek A, Pötschger U, Kastner U, Flege S, Kempf-Bielack B, et al. Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 2003;21:2011–8. - PubMed
-
- Kim MS, Cho WH, Song WS, Lee SY, Jeon DG. Time dependency of prognostic factors in patients with stage II osteosarcomas. Clin Orthop Relat Res 2007;463:157–65. - PubMed
-
- Bielack SS, Kempf-Bielack B, Delling G, Exner GU, Flege S, Helmke K, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 2002;20:776–90. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
