

Systematic identification of gene combinations to target in innate immune cells to enhance T cell activation
- PMID: 37813864
- PMCID: PMC10562403
- DOI: 10.1038/s41467-023-41792-8
Systematic identification of gene combinations to target in innate immune cells to enhance T cell activation
Abstract
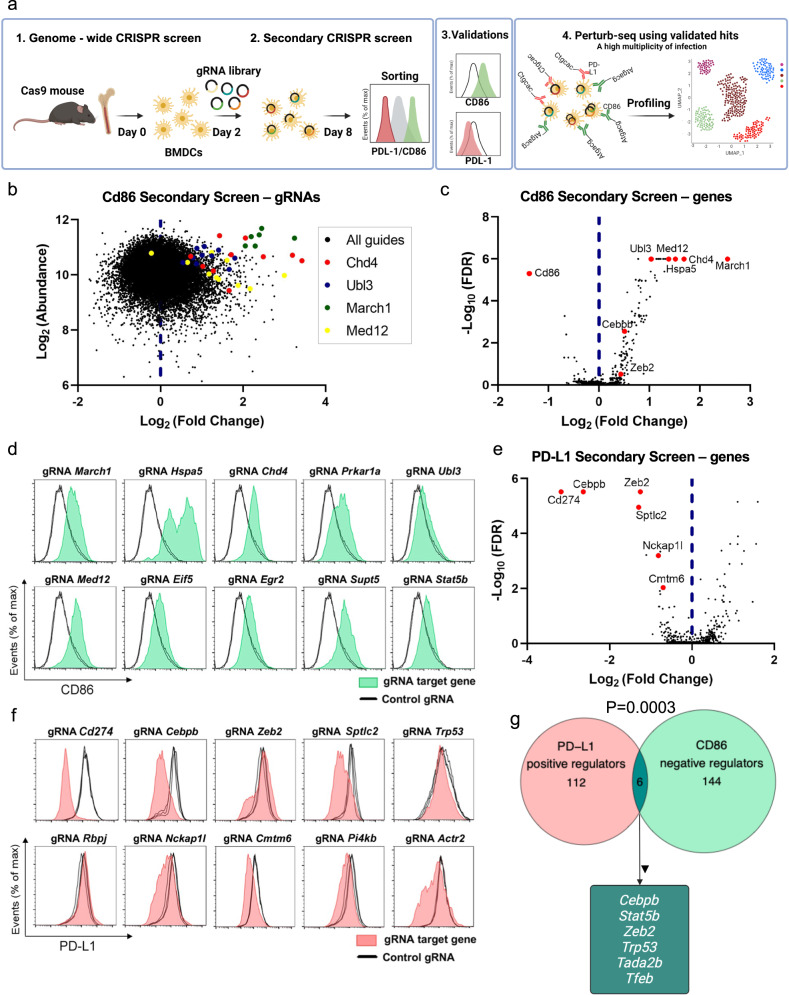
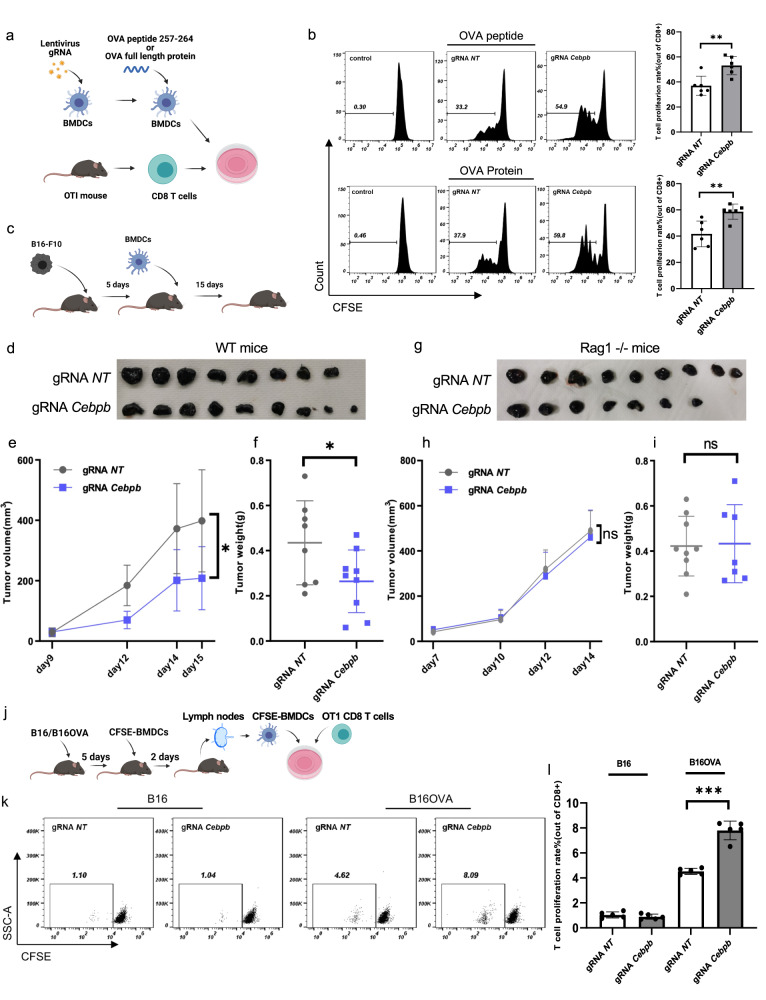
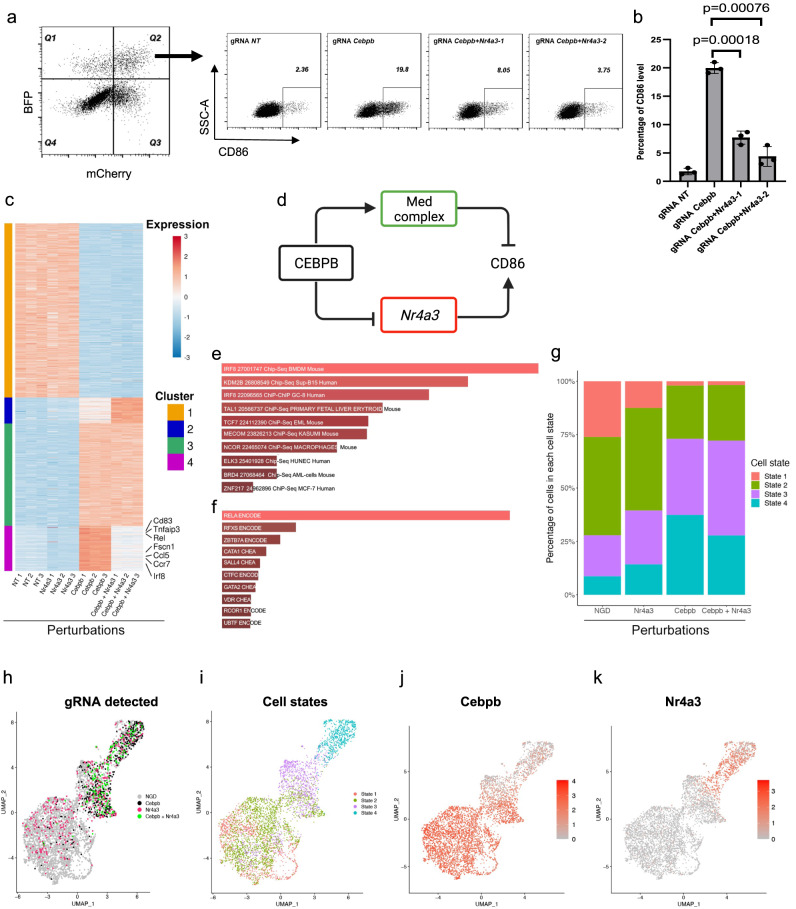
Genetic engineering of immune cells has opened new avenues for improving their functionality but it remains a challenge to pinpoint which genes or combination of genes are the most beneficial to target. Here, we conduct High Multiplicity of Perturbations and Cellular Indexing of Transcriptomes and Epitopes (HMPCITE-seq) to find combinations of genes whose joint targeting improves antigen-presenting cell activity and enhances their ability to activate T cells. Specifically, we perform two genome-wide CRISPR screens in bone marrow dendritic cells and identify negative regulators of CD86, that participate in the co-stimulation programs, including Chd4, Stat5b, Egr2, Med12, and positive regulators of PD-L1, that participate in the co-inhibitory programs, including Sptlc2, Nckap1l, and Pi4kb. To identify the genetic interactions between top-ranked genes and find superior combinations to target, we perform high-order Perturb-Seq experiments and we show that targeting both Cebpb and Med12 results in a better phenotype compared to the single perturbations or other combinations of perturbations.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interest.
Figures



References
-
- Lu Y, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020;26:732–740. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
