

Rapid discovery of high-affinity antibodies via massively parallel sequencing, ribosome display and affinity screening
- PMID: 37814006
- PMCID: PMC10963267
- DOI: 10.1038/s41551-023-01093-3
Rapid discovery of high-affinity antibodies via massively parallel sequencing, ribosome display and affinity screening
Abstract
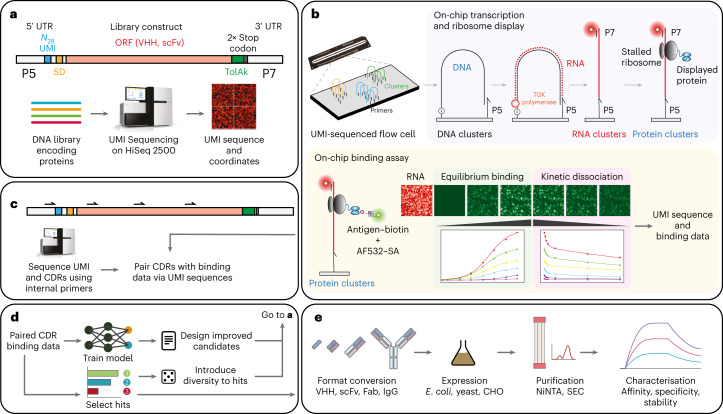
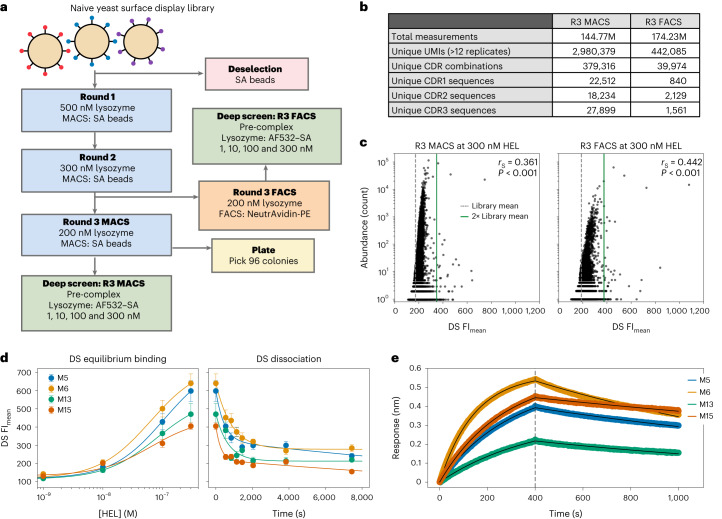
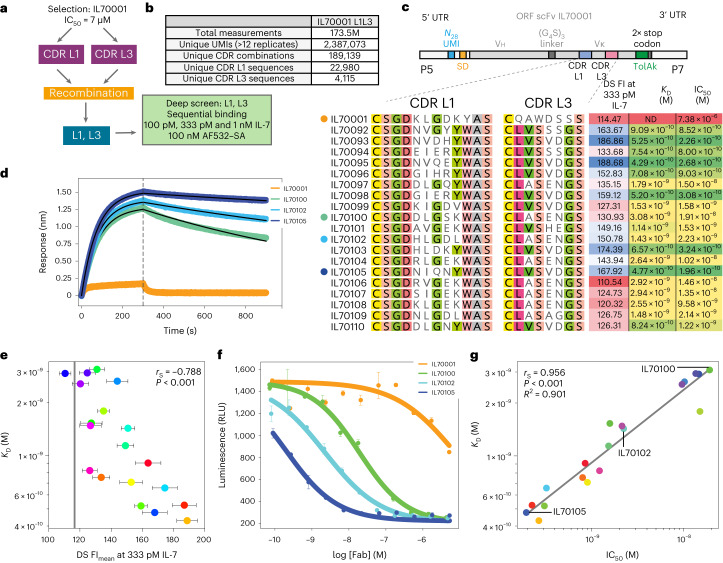
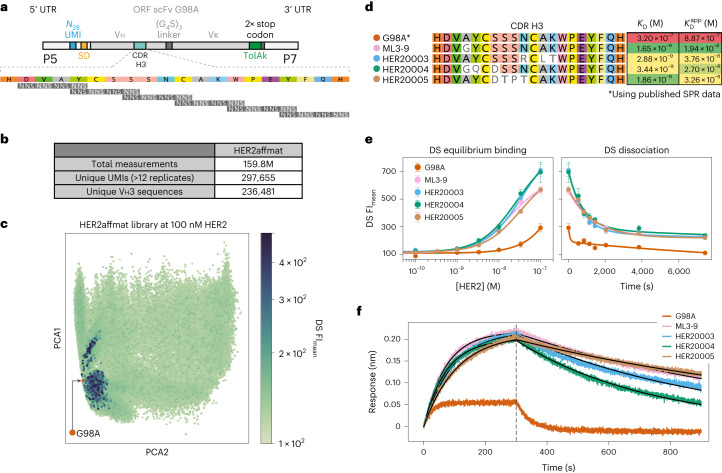
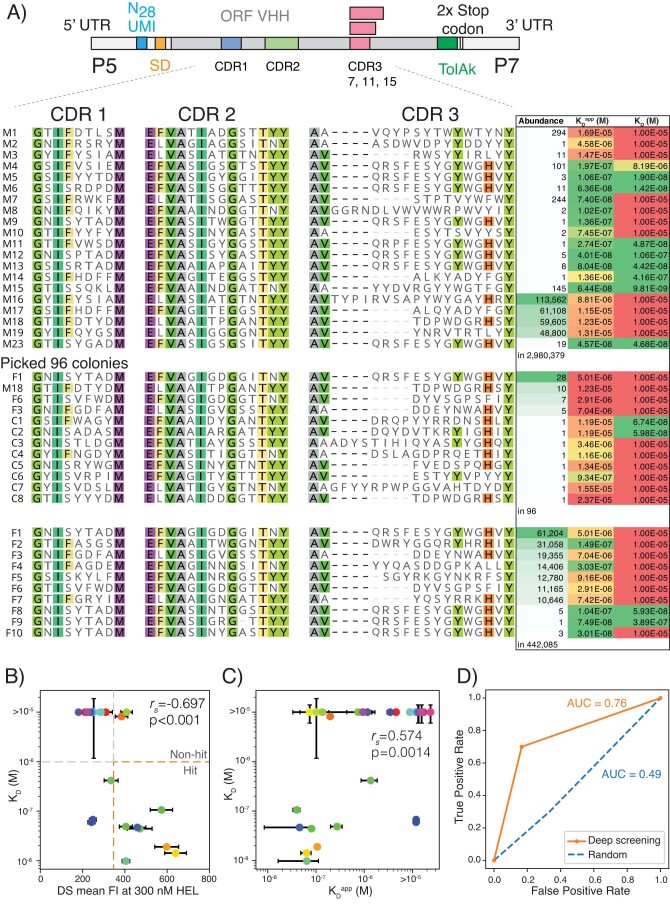
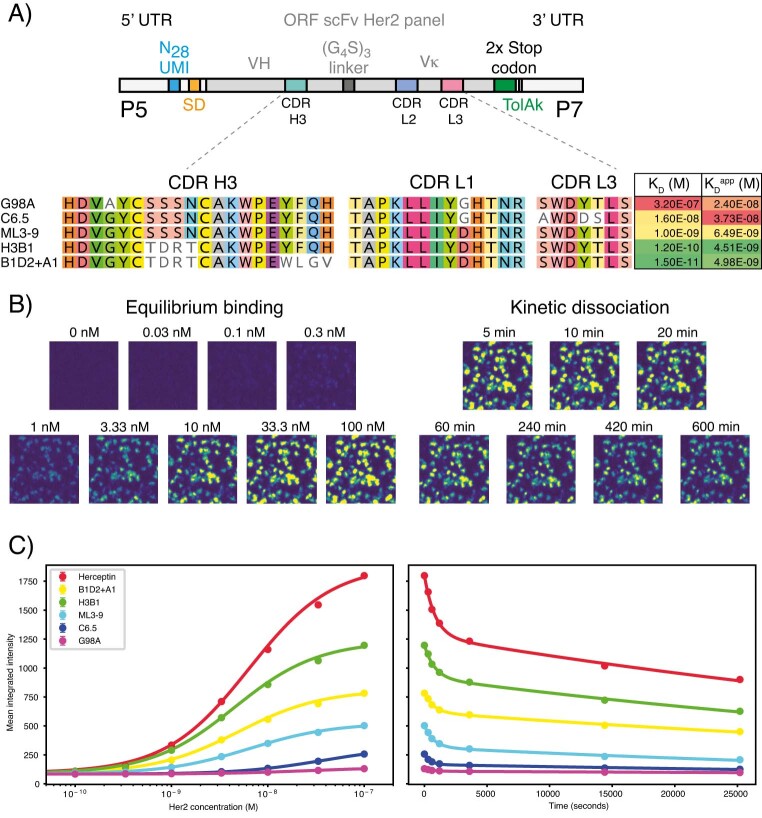
Developing therapeutic antibodies is laborious and costly. Here we report a method for antibody discovery that leverages the Illumina HiSeq platform to, within 3 days, screen in the order of 108 antibody-antigen interactions. The method, which we named 'deep screening', involves the clustering and sequencing of antibody libraries, the conversion of the DNA clusters into complementary RNA clusters covalently linked to the instrument's flow-cell surface on the same location, the in situ translation of the clusters into antibodies tethered via ribosome display, and their screening via fluorescently labelled antigens. By using deep screening, we discovered low-nanomolar nanobodies to a model antigen using 4 × 106 unique variants from yeast-display-enriched libraries, and high-picomolar single-chain antibody fragment leads for human interleukin-7 directly from unselected synthetic repertoires. We also leveraged deep screening of a library of 2.4 × 105 sequences of the third complementarity-determining region of the heavy chain of an anti-human epidermal growth factor receptor 2 (HER2) antibody as input for a large language model that generated new single-chain antibody fragment sequences with higher affinity for HER2 than those in the original library.
© 2023. The Author(s).
Conflict of interest statement
The MRC-LMB has filed a patent application on the methodologies described in this article, with B.T.P. and P.H. named as inventors. A.B., G.B. and A.R. are current employees of AstraZeneca. M.B. is a current employee of UCB Pharma. R.M. is a current employee of Alchemab Therapeutics. The other authors declare no competing interests.
Figures




References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
