

Senescence, brain inflammation, and oligomeric tau drive cognitive decline in Alzheimer's disease: Evidence from clinical and preclinical studies
- PMID: 37814508
- PMCID: PMC10841264
- DOI: 10.1002/alz.13490
Senescence, brain inflammation, and oligomeric tau drive cognitive decline in Alzheimer's disease: Evidence from clinical and preclinical studies
Abstract
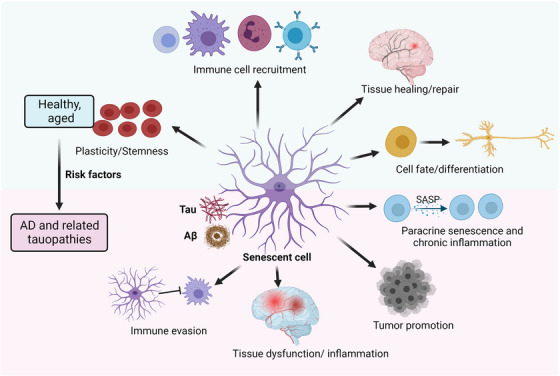
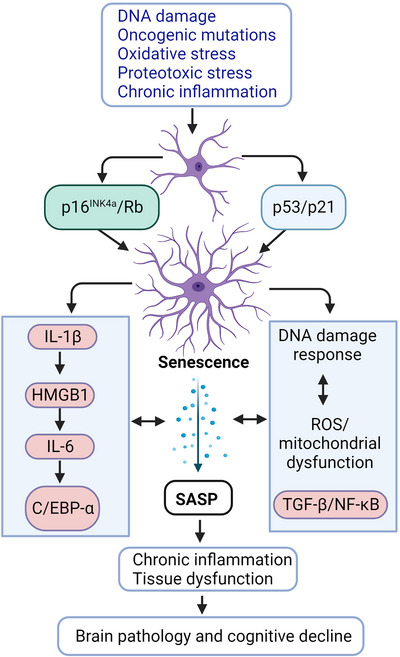
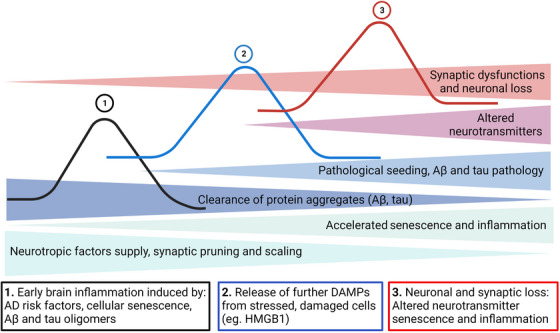
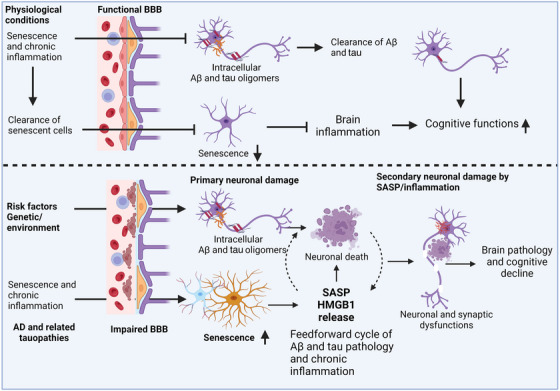
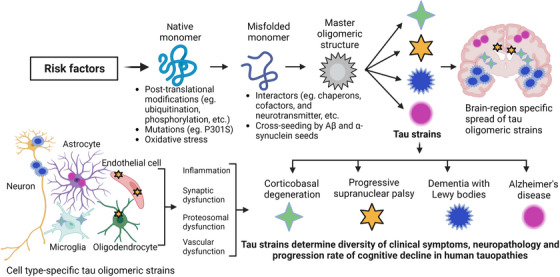
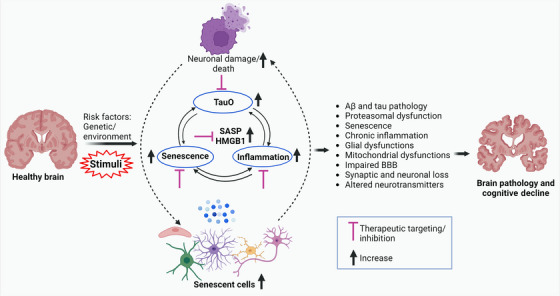
Aging, tau pathology, and chronic inflammation in the brain play crucial roles in synaptic loss, neurodegeneration, and cognitive decline in tauopathies, including Alzheimer's disease. Senescent cells accumulate in the aging brain, accelerate the aging process, and promote tauopathy progression through their abnormal inflammatory secretome known as the senescence-associated secretory phenotype (SASP). Tau oligomers (TauO)-the most neurotoxic tau species-are known to induce senescence and the SASP, which subsequently promote neuropathology, inflammation, oxidative stress, synaptic dysfunction, neuronal death, and cognitive dysfunction. TauO, brain inflammation, and senescence are associated with heterogeneity in tauopathy progression and cognitive decline. However, the underlying mechanisms driving the disease heterogeneity remain largely unknown, impeding the development of therapies for tauopathies. Based on clinical and preclinical evidence, this review highlights the critical role of TauO and senescence in neurodegeneration. We discuss key knowledge gaps and potential strategies for targeting senescence and TauO to treat tauopathies. HIGHLIGHTS: Senescence, oligomeric Tau (TauO), and brain inflammation accelerate the aging process and promote the progression of tauopathies, including Alzheimer's disease. We discuss their role in contributing to heterogeneity in tauopathy and cognitive decline. We highlight strategies to target senescence and TauO to treat tauopathies while addressing key knowledge gaps.
Keywords: Alzheimer's disease; aging; cognitive deficits; inflammation; senescence-associated secretory phenotype; senescent cells; tau; tauopathies.
© 2023 the Alzheimer's Association.
Conflict of interest statement
The authors declare that there are no conflicts of financial interest related to this publication. Author disclosures are available in the supporting information.
Figures
References
-
- Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631‐639. - PubMed
-
- Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495‐1500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
