

An Influenza A virus can evolve to use human ANP32E through altering polymerase dimerization
- PMID: 37816726
- PMCID: PMC10564888
- DOI: 10.1038/s41467-023-41308-4
An Influenza A virus can evolve to use human ANP32E through altering polymerase dimerization
Abstract
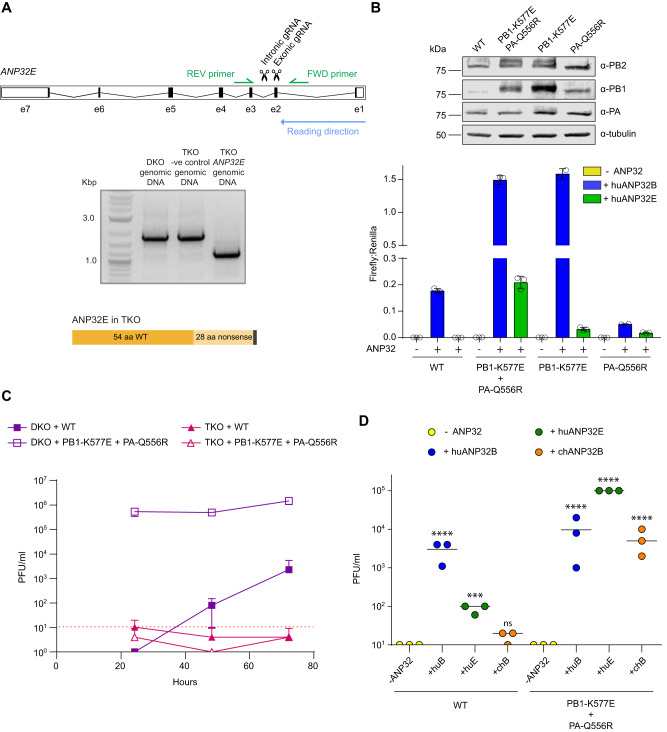
Human ANP32A and ANP32B are essential but redundant host factors for influenza virus genome replication. While most influenza viruses cannot replicate in edited human cells lacking both ANP32A and ANP32B, some strains exhibit limited growth. Here, we experimentally evolve such an influenza A virus in these edited cells and unexpectedly, after 2 passages, we observe robust viral growth. We find two mutations in different subunits of the influenza polymerase that enable the mutant virus to use a novel host factor, ANP32E, an alternative family member, which is unable to support the wild type polymerase. Both mutations reside in the symmetric dimer interface between two polymerase complexes and reduce polymerase dimerization. These mutations have previously been identified as adapting influenza viruses to mice. Indeed, the evolved virus gains the ability to use suboptimal mouse ANP32 proteins and becomes more virulent in mice. We identify further mutations in the symmetric dimer interface which we predict allow influenza to adapt to use suboptimal ANP32 proteins through a similar mechanism. Overall, our results suggest a balance between asymmetric and symmetric dimers of influenza virus polymerase that is influenced by the interaction between polymerase and ANP32 host proteins.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
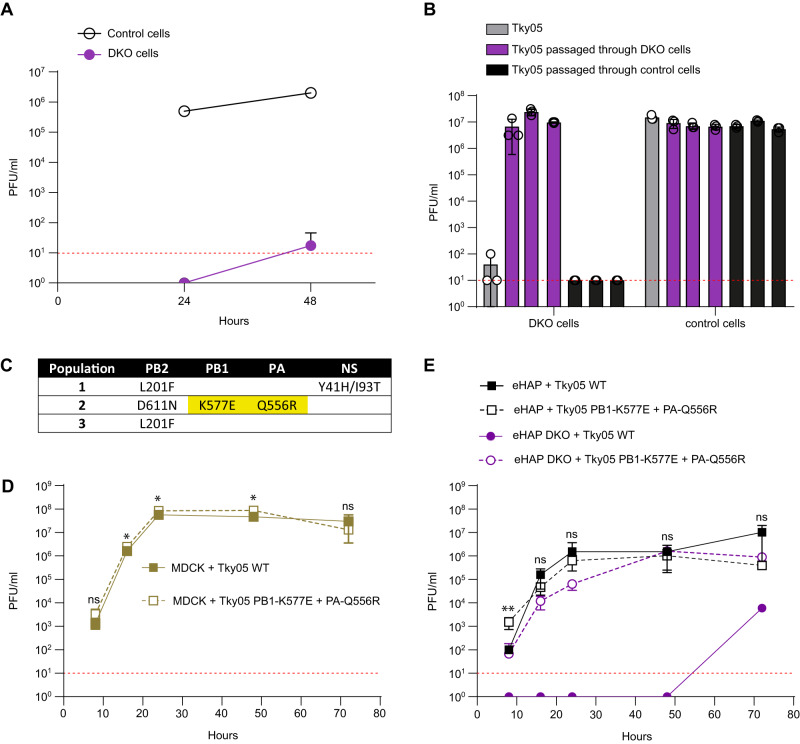
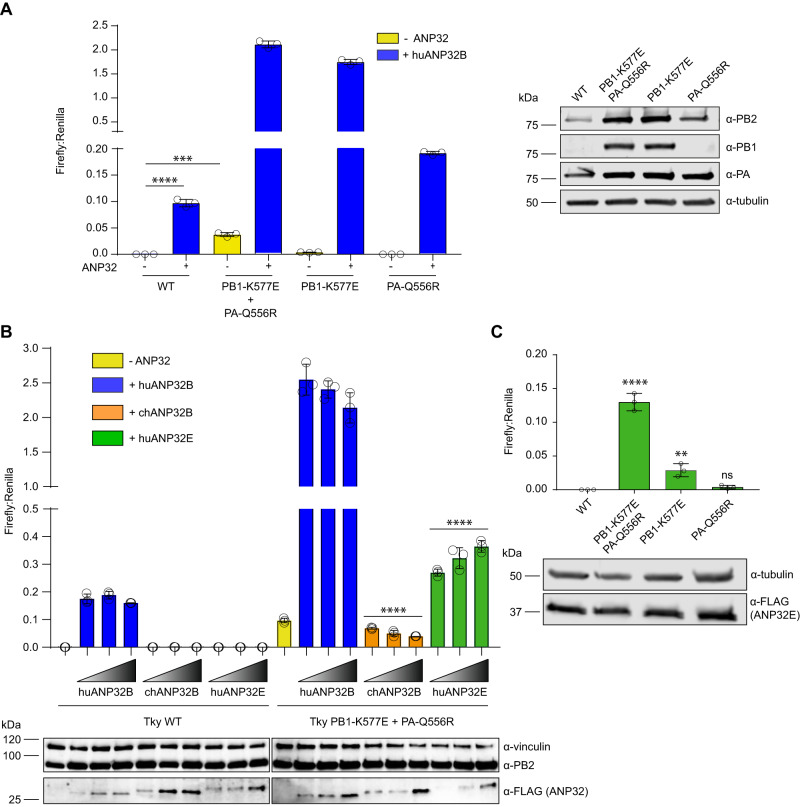
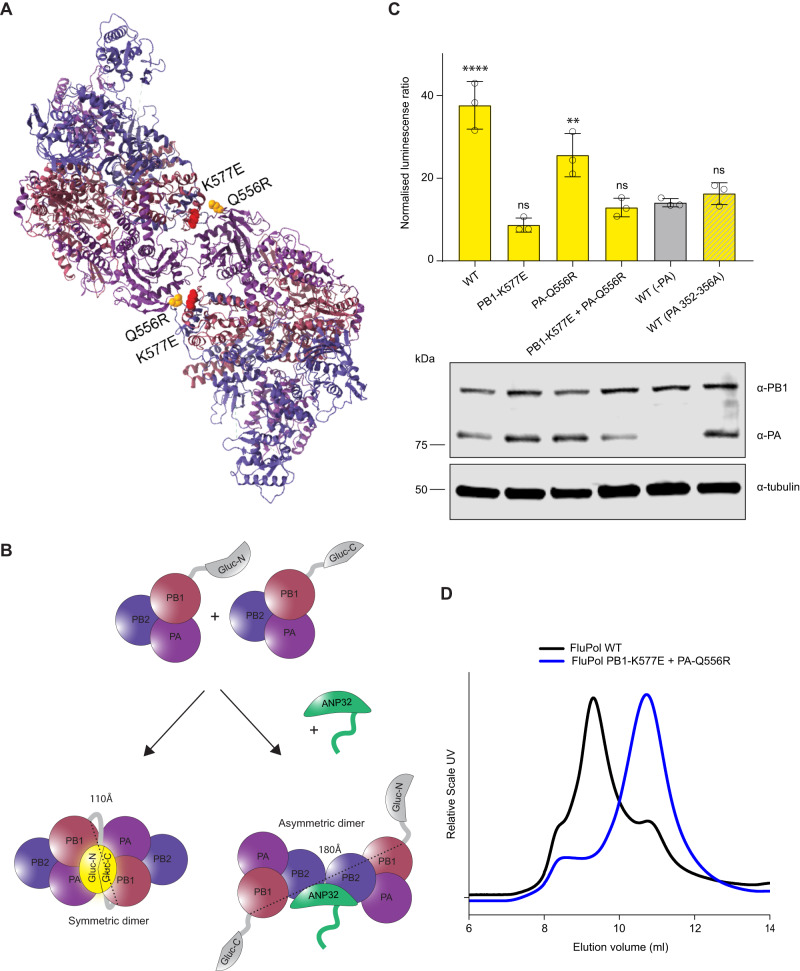
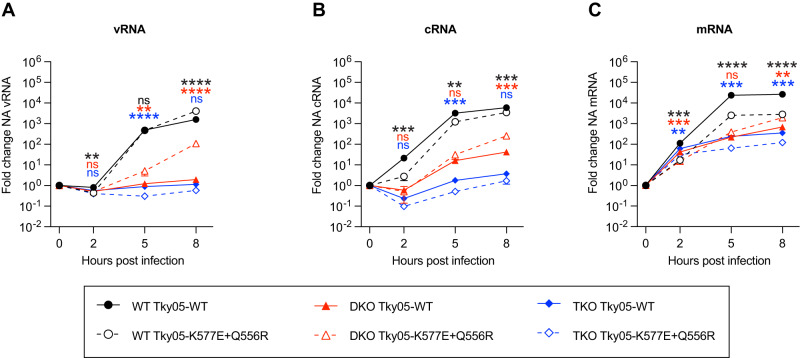
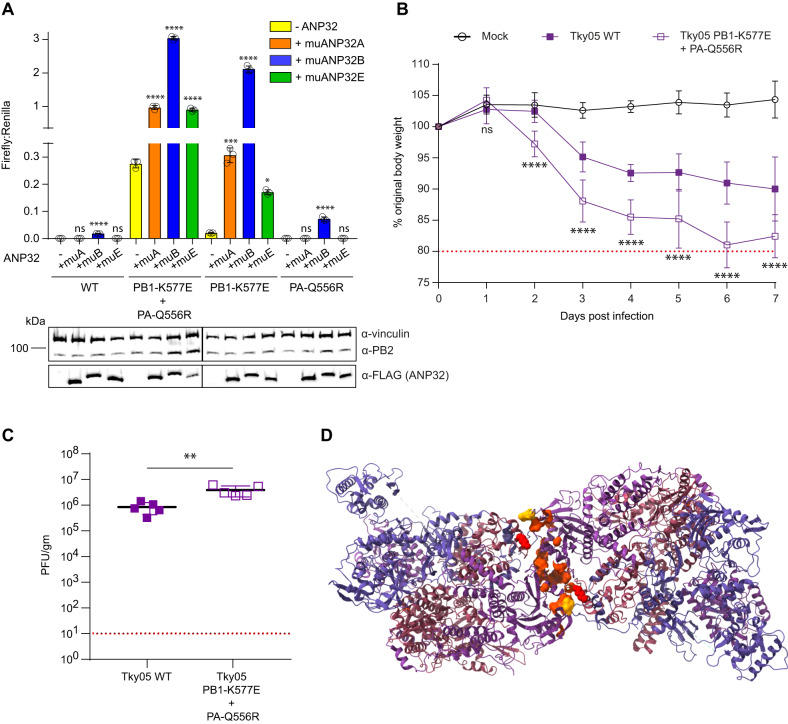
References
Publication types
MeSH terms
Substances
Grants and funding
- BB/R007292/1 /BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 205100/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MR/R009945/1/MRC_/Medical Research Council/United Kingdom
- BB/R013071/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 205100/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
