

Multimodal analysis of methylomics and fragmentomics in plasma cell-free DNA for multi-cancer early detection and localization
- PMID: 37819044
- PMCID: PMC10567114
- DOI: 10.7554/eLife.89083
Multimodal analysis of methylomics and fragmentomics in plasma cell-free DNA for multi-cancer early detection and localization
Abstract
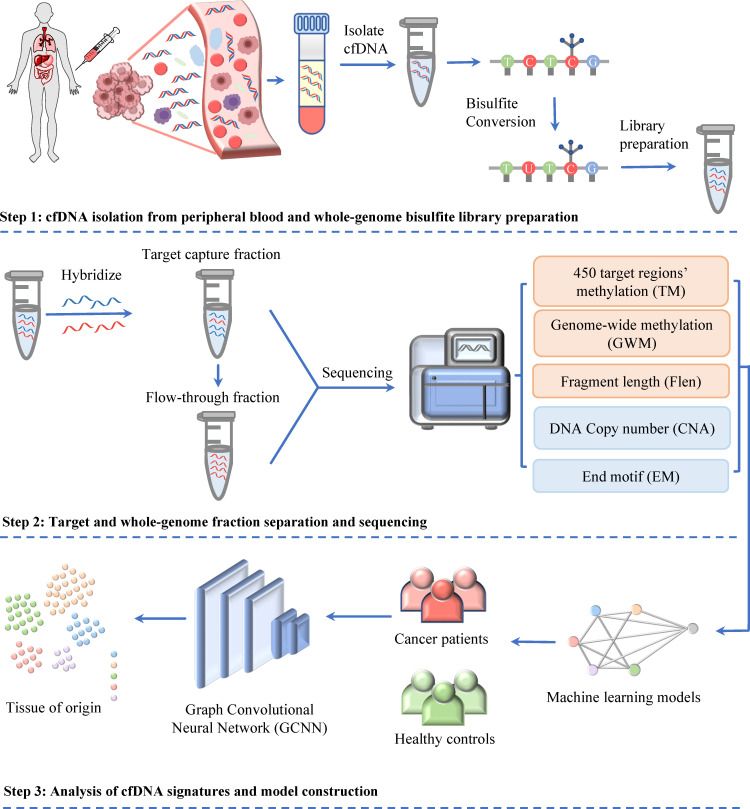
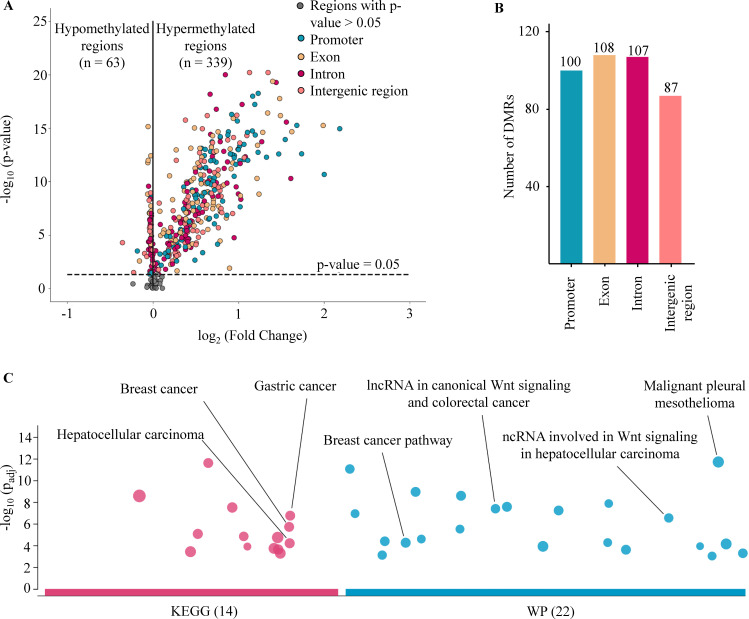
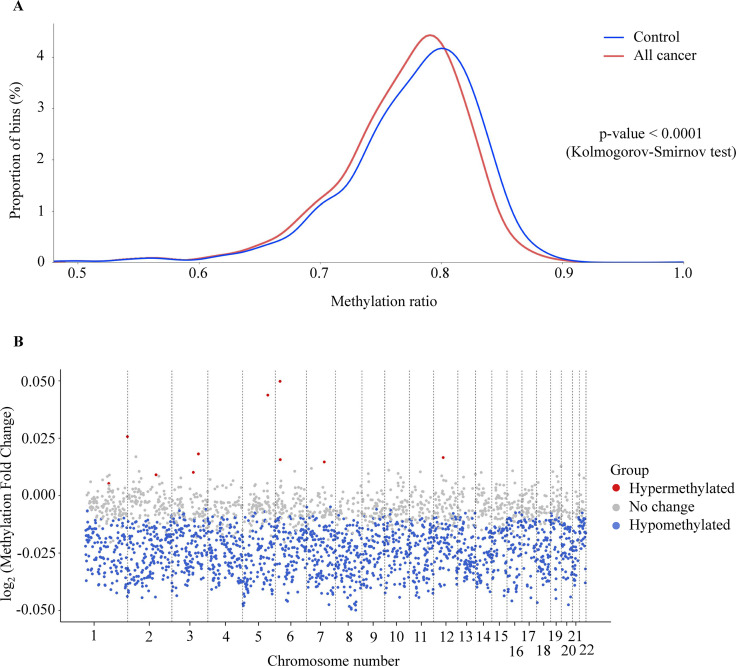
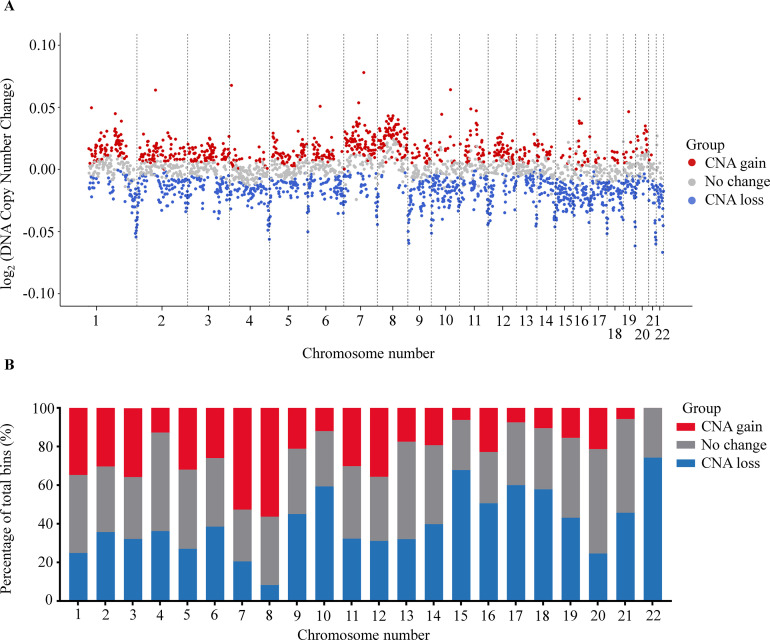
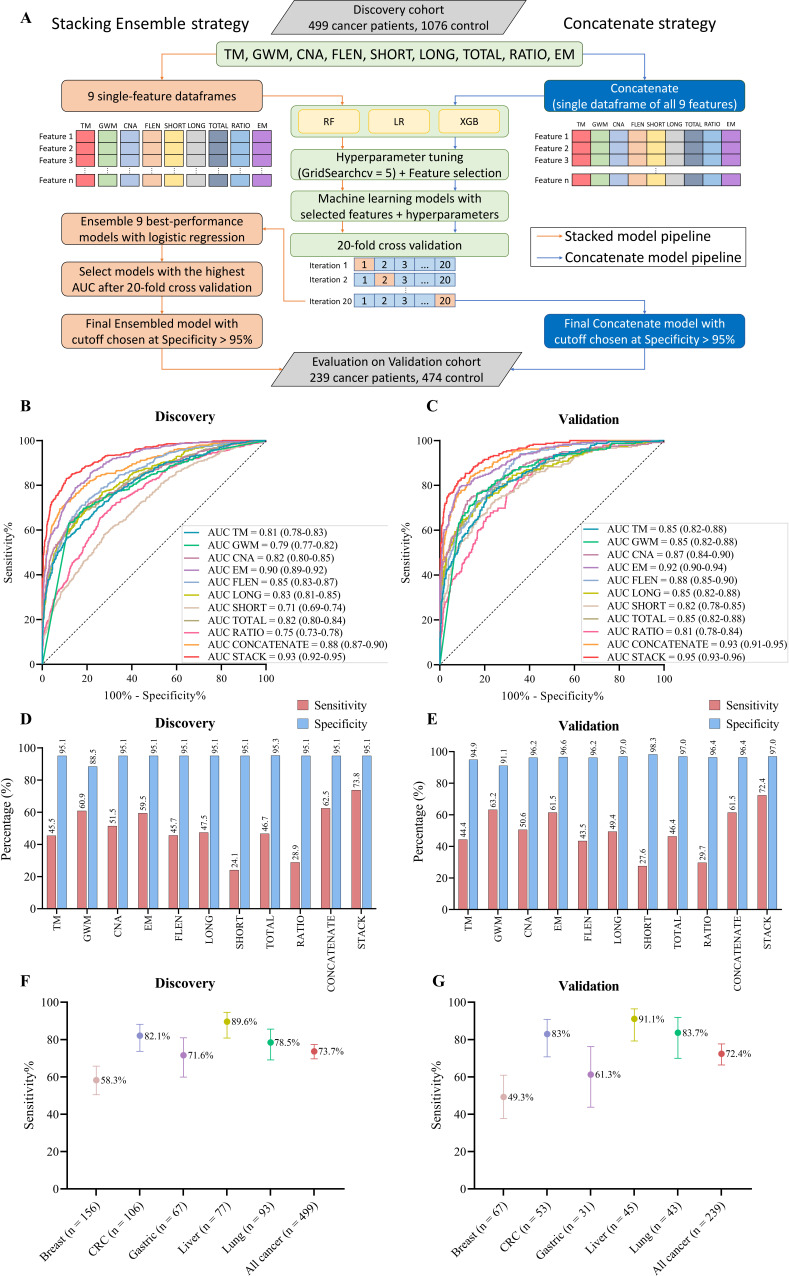
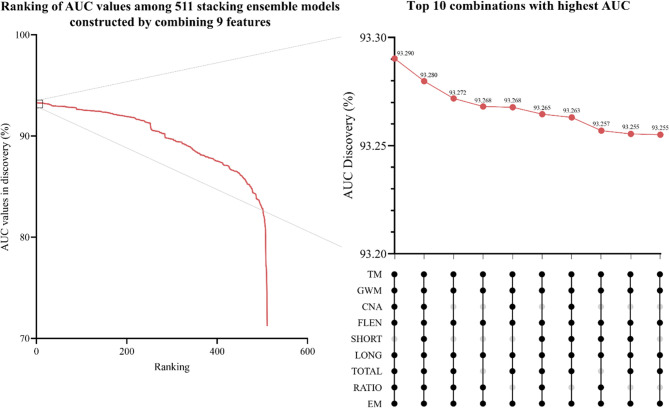
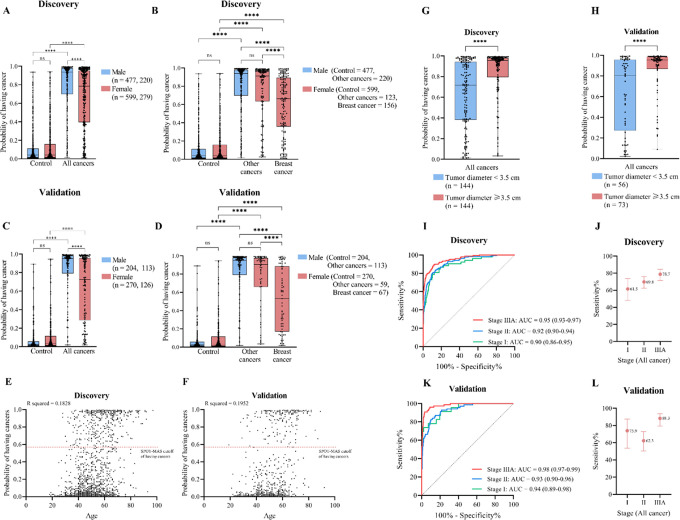
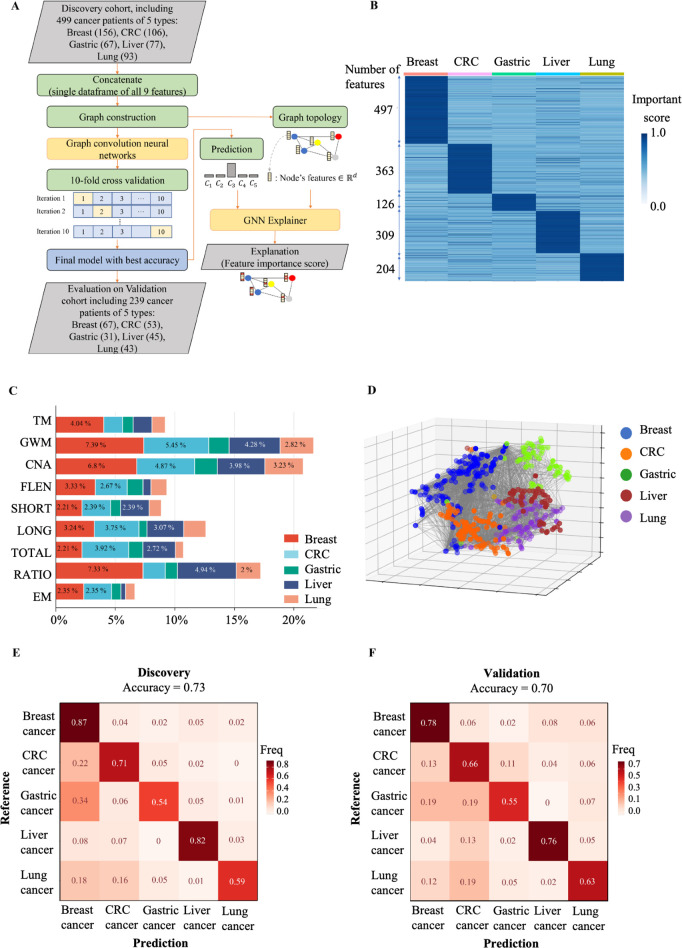
Despite their promise, circulating tumor DNA (ctDNA)-based assays for multi-cancer early detection face challenges in test performance, due mostly to the limited abundance of ctDNA and its inherent variability. To address these challenges, published assays to date demanded a very high-depth sequencing, resulting in an elevated price of test. Herein, we developed a multimodal assay called SPOT-MAS (screening for the presence of tumor by methylation and size) to simultaneously profile methylomics, fragmentomics, copy number, and end motifs in a single workflow using targeted and shallow genome-wide sequencing (~0.55×) of cell-free DNA. We applied SPOT-MAS to 738 non-metastatic patients with breast, colorectal, gastric, lung, and liver cancer, and 1550 healthy controls. We then employed machine learning to extract multiple cancer and tissue-specific signatures for detecting and locating cancer. SPOT-MAS successfully detected the five cancer types with a sensitivity of 72.4% at 97.0% specificity. The sensitivities for detecting early-stage cancers were 73.9% and 62.3% for stages I and II, respectively, increasing to 88.3% for non-metastatic stage IIIA. For tumor-of-origin, our assay achieved an accuracy of 0.7. Our study demonstrates comparable performance to other ctDNA-based assays while requiring significantly lower sequencing depth, making it economically feasible for population-wide screening.
Keywords: cancer biology; circulating tumor DNA; genetics; genomics; human; liquid biopsy; multimodal analysis.
© 2023, Nguyen, Nguyen et al.
Conflict of interest statement
VN VTCN is affiliated with Gene Solutions. The author has no other competing interests to declare, TN HTN is affiliated with Gene Solutions. The author has no other competing interests to declare, ND NNTD is affiliated with Gene Solutions. The author has no other competing interests to declare, TP TMQP is affiliated with Gene Solutions. The author has no other competing interests to declare, GN GTHN is affiliated with Gene Solutions. The author has no other competing interests to declare, TN TDN is affiliated with Gene Solutions. The author has no other competing interests to declare, TT TTTT is affiliated with Gene Solutions. The author has no other competing interests to declare, DV, TP, TJ, VN, HN, TN, QD, TD, AT, VN, VN, LH, QT, TP, TH, BN, TN, TN, DP, BP, TV, TN, TT, MT, NT, TL, TT, MD, HB, VK, TP, DT, TL, TP, ML, VC, TD No competing interests declared, TN THHN is affiliated with Gene Solutions. The author has no other competing interests to declare, LH LAKH is affiliated with Gene Solutions. The author has no other competing interests to declare, TT THT is affiliated with Gene Solutions. The author has no other competing interests to declare, DV DHV is affiliated with Gene Solutions. The author has no other competing interests to declare, TT TMTT is affiliated with Gene Solutions. The author has no other competing interests to declare, MN MNN is affiliated with Gene Solutions. The author has no other competing interests to declare, TV TTVV is affiliated with Gene Solutions. The author has no other competing interests to declare, AN ANN is affiliated with Gene Solutions. The author has no other competing interests to declare, TT TTT is affiliated with Gene Solutions. The author has no other competing interests to declare, VT VUT is affiliated with Gene Solutions. The author has no other competing interests to declare, ML MPL is affiliated with Gene Solutions. The author has no other competing interests to declare, TD TTD is affiliated with Gene Solutions. The author has no other competing interests to declare, TP TVP is affiliated with Gene Solutions. The author has no other competing interests to declare, HN HDN is affiliated with Gene Solutions. The author has no other competing interests to declare, DN DSN holds equity in Gene Solutions.DSN is affiliated with Gene Solutions. The author has no other competing interests to declare, DT DKT is affiliated with Gene Solutions. The author has no other competing interests to declare, HT HST is affiliated with Gene Solutions. The author has no other competing interests to declare, HG HG holds equity in Gene Solutions. The funder Gene Solutions provided support in the form of salaries for HG who is inventor on the patent application (USPTO 17930705).HG is affiliated with Gene Solutions. The author has no other competing interests to declare, HN HNN holds equity in Gene Solutions. The funder Gene Solutions provided support in the form of salaries for HNN who is inventor on the patent application (USPTO 17930705).HNN is affiliated with Gene Solutions. The author has no other competing interests to declare, MP MDP holds equity in Gene Solutions. The funder Gene Solutions provided support in the form of salaries for MDP who is inventor on the patent application (USPTO 17930705).MDP is affiliated with Gene Solutions. The author has no other competing interests to declare, LT LST holds equity in Gene Solutions. The funder Gene Solutions provided support in the form of salaries for LST who is inventor on the patent application (USPTO 17930705).LST is affiliated with Gene Solutions. The author has no other competing interests to declare
Figures
Update of
- doi: 10.1101/2023.04.12.23288460
- doi: 10.7554/eLife.89083.1
- doi: 10.7554/eLife.89083.2
References
-
- Chan KCA, Jiang P, Chan CWM, Sun K, Wong J, Hui EP, Chan SL, Chan WC, Hui DSC, Ng SSM, Chan HLY, Wong CSC, Ma BBY, Chan ATC, Lai PBS, Sun H, Chiu RWK, Lo YMD. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. PNAS. 2013;110:18761–18768. doi: 10.1073/pnas.1313995110. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
