

Learning from prepandemic data to forecast viral escape
- PMID: 37821700
- PMCID: PMC10599991
- DOI: 10.1038/s41586-023-06617-0
Learning from prepandemic data to forecast viral escape
Abstract
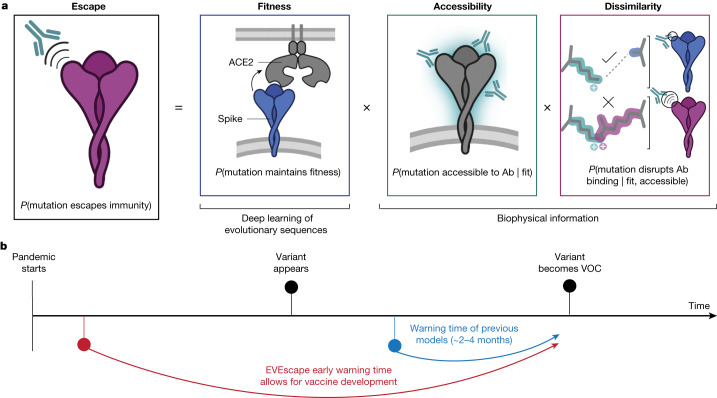
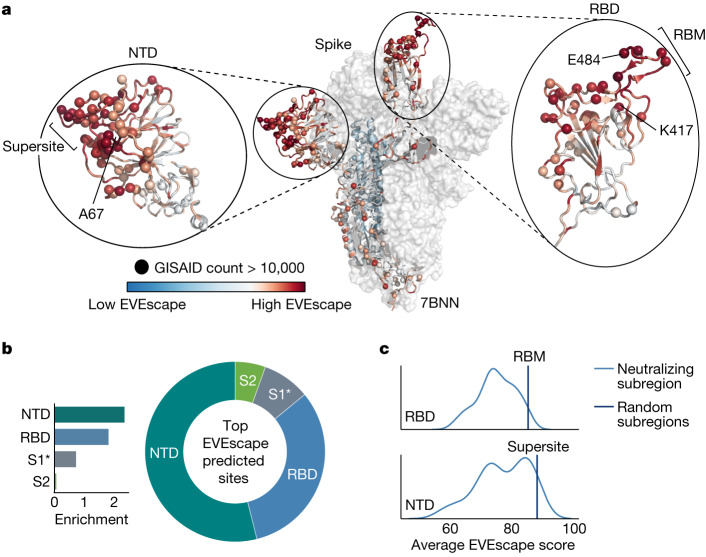
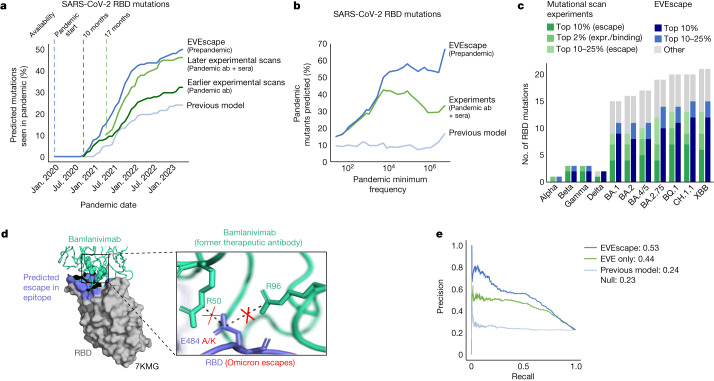
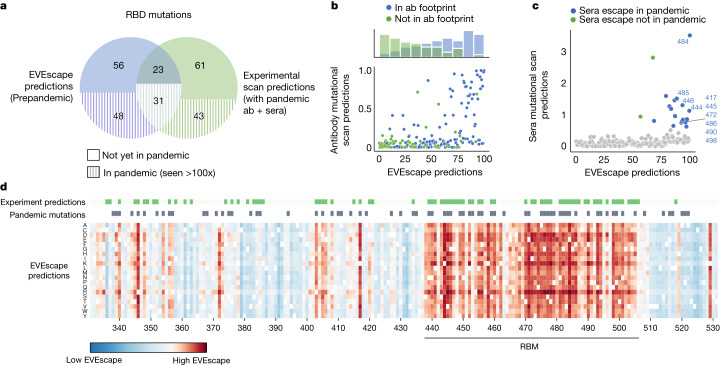
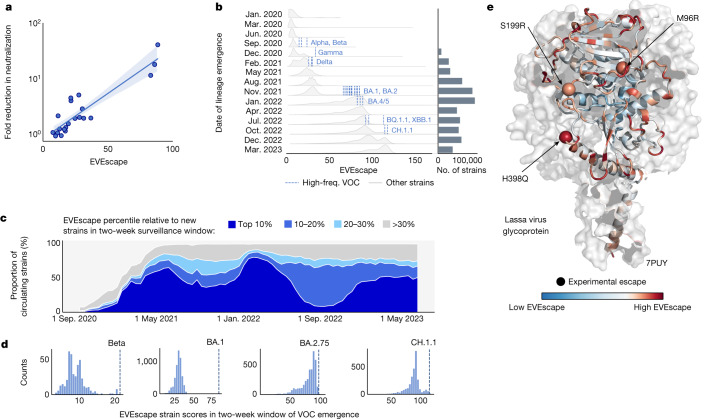
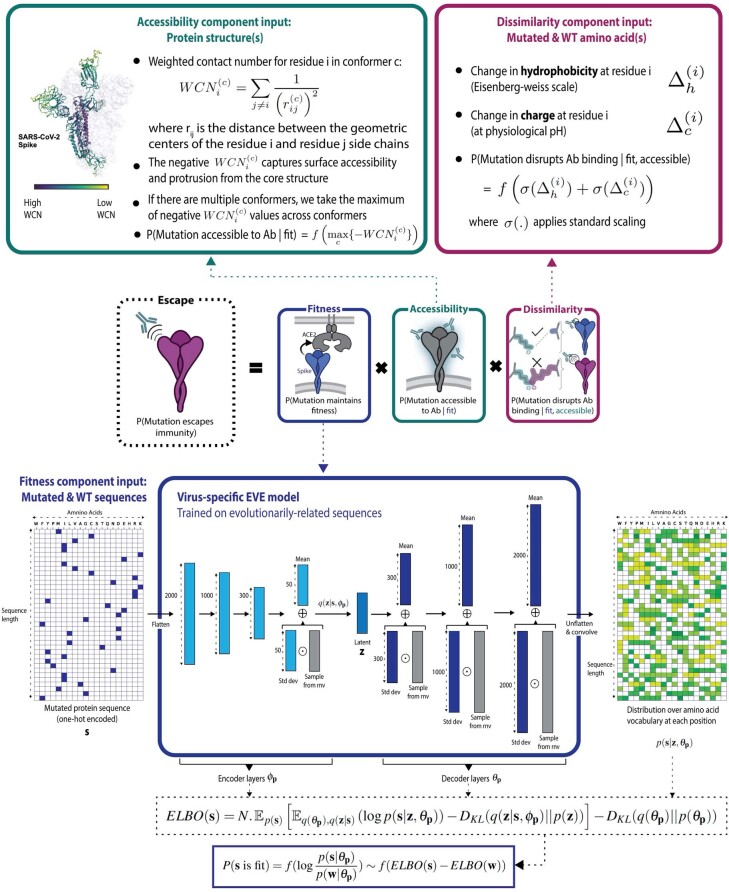
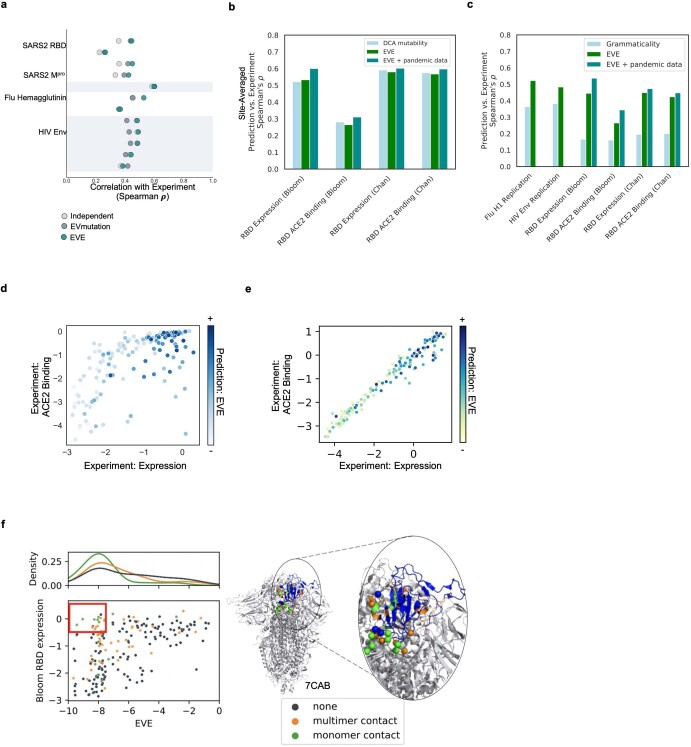
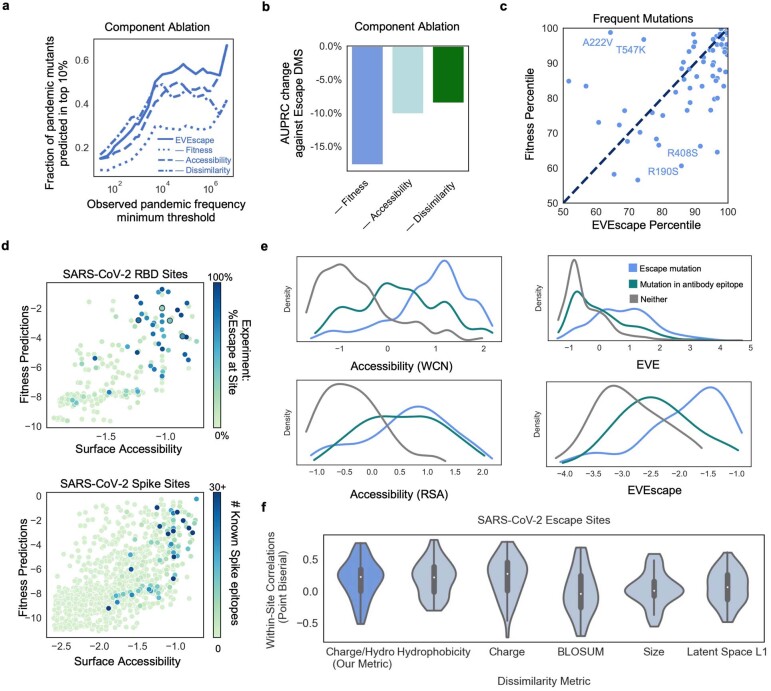
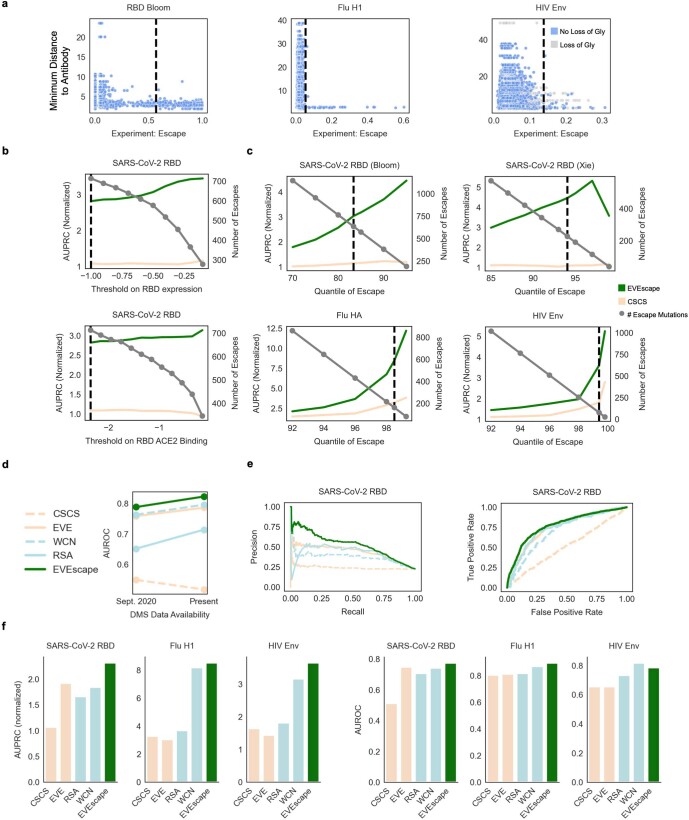
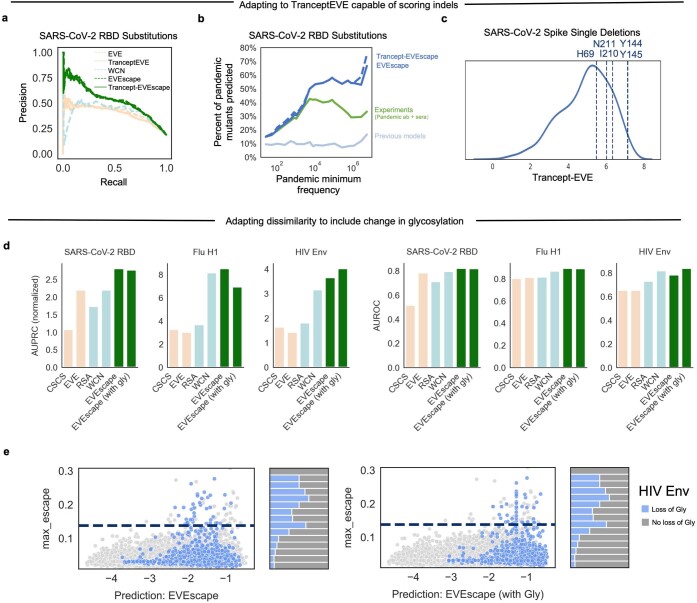
Effective pandemic preparedness relies on anticipating viral mutations that are able to evade host immune responses to facilitate vaccine and therapeutic design. However, current strategies for viral evolution prediction are not available early in a pandemic-experimental approaches require host polyclonal antibodies to test against1-16, and existing computational methods draw heavily from current strain prevalence to make reliable predictions of variants of concern17-19. To address this, we developed EVEscape, a generalizable modular framework that combines fitness predictions from a deep learning model of historical sequences with biophysical and structural information. EVEscape quantifies the viral escape potential of mutations at scale and has the advantage of being applicable before surveillance sequencing, experimental scans or three-dimensional structures of antibody complexes are available. We demonstrate that EVEscape, trained on sequences available before 2020, is as accurate as high-throughput experimental scans at anticipating pandemic variation for SARS-CoV-2 and is generalizable to other viruses including influenza, HIV and understudied viruses with pandemic potential such as Lassa and Nipah. We provide continually revised escape scores for all current strains of SARS-CoV-2 and predict probable further mutations to forecast emerging strains as a tool for continuing vaccine development ( evescape.org ).
© 2023. The Author(s).
Conflict of interest statement
D.S.M. is an advisor for Dyno Therapeutics, Octant, Jura Bio, Tectonic Therapeutic and Genentech and is a cofounder of Seismic Therapeutic. C.S. is an advisor for CytoReason Ltd. The remaining authors declare no competing interests.
Figures
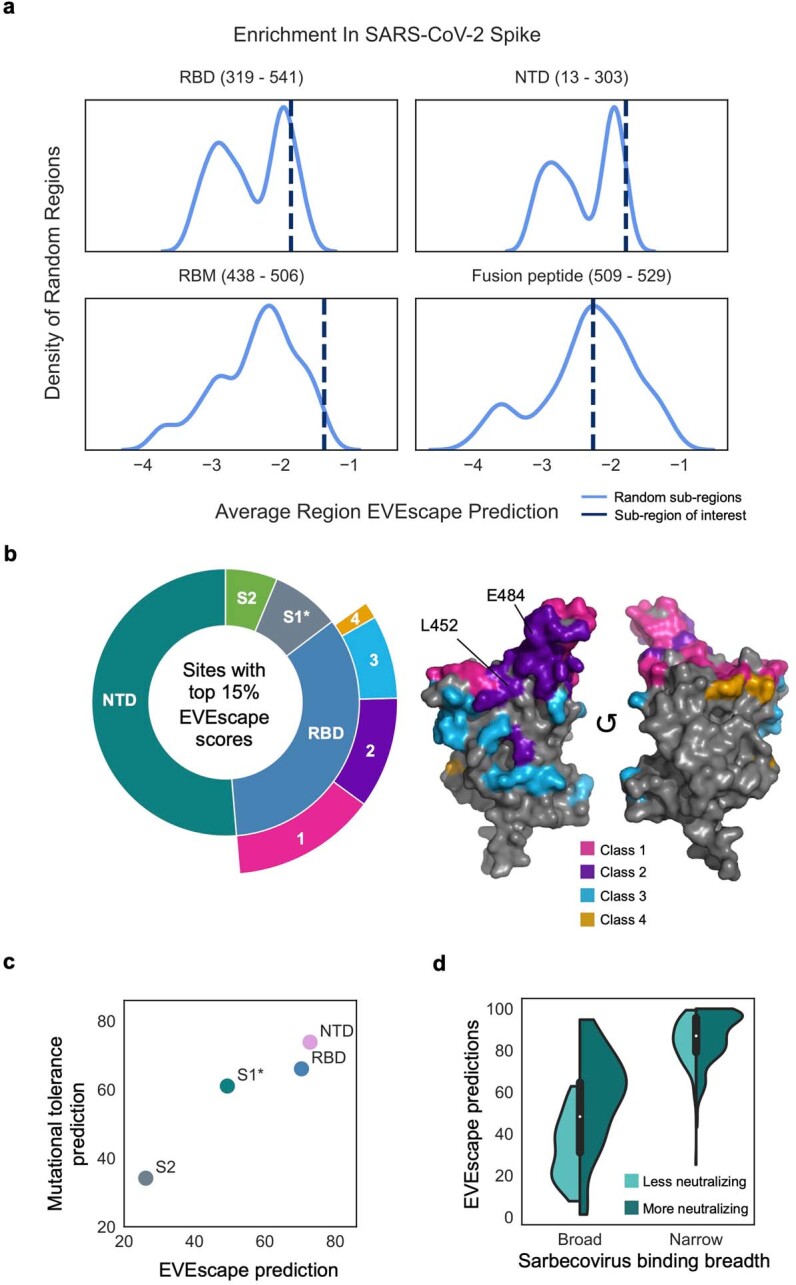
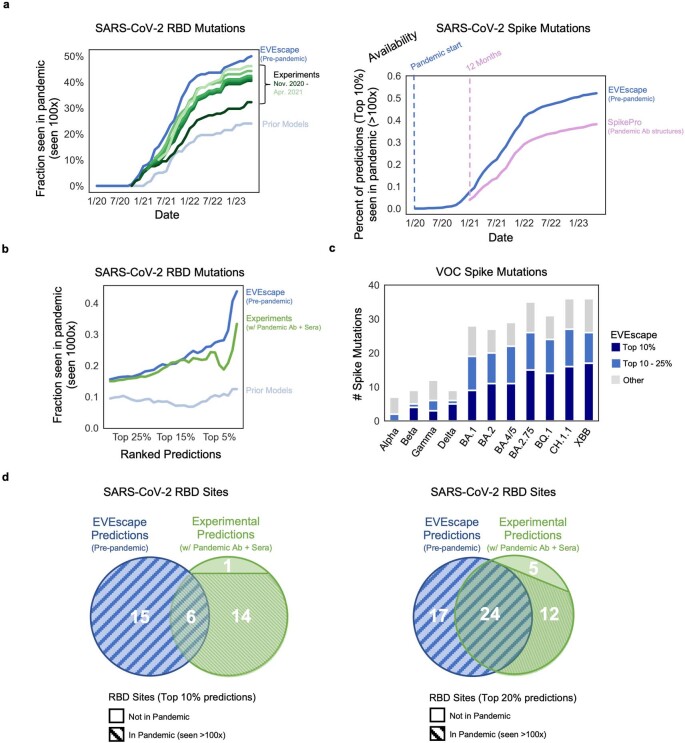
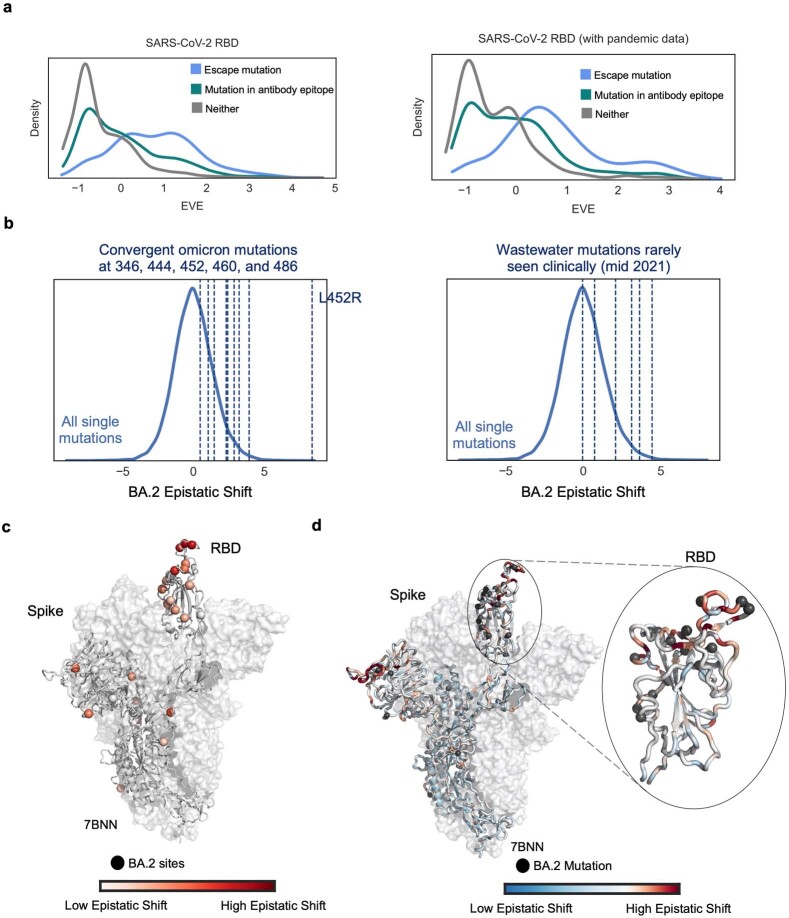
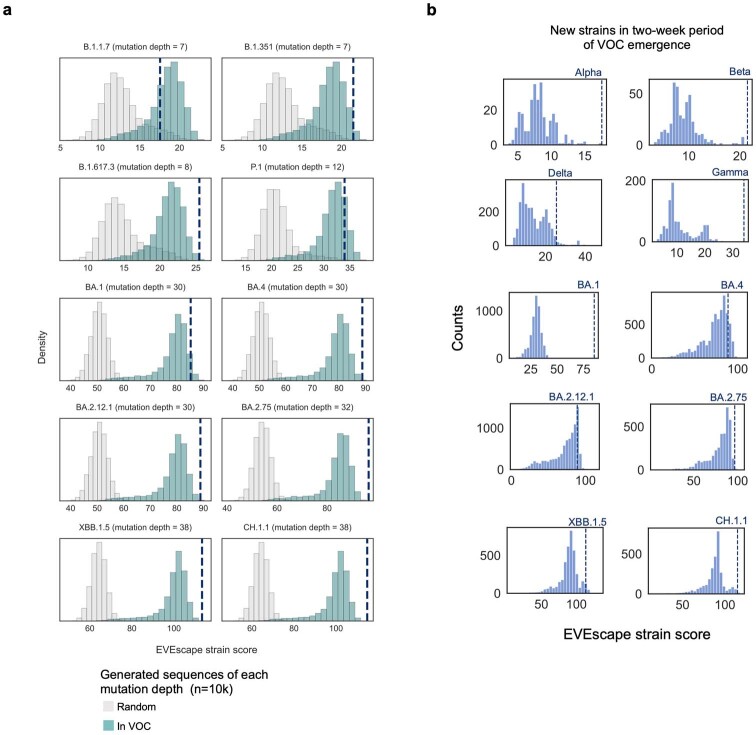
Comment in
-
Learn from the past to predict viral pandemics.Nature. 2023 Oct;622(7984):700-702. doi: 10.1038/d41586-023-02931-9. Nature. 2023. PMID: 37821603 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
