

Mexican Biobank advances population and medical genomics of diverse ancestries
- PMID: 37821706
- PMCID: PMC10600006
- DOI: 10.1038/s41586-023-06560-0
Mexican Biobank advances population and medical genomics of diverse ancestries
Abstract
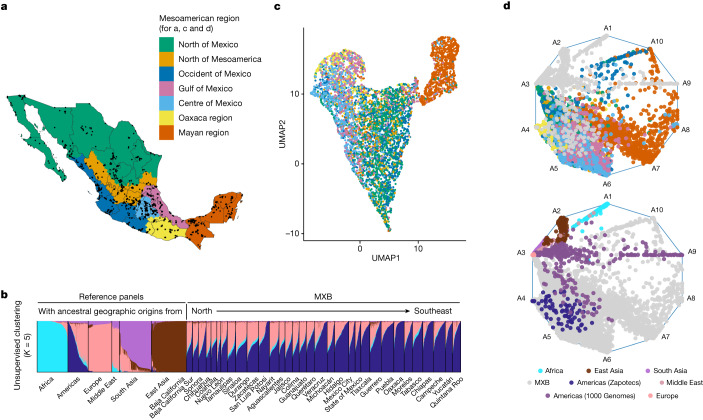
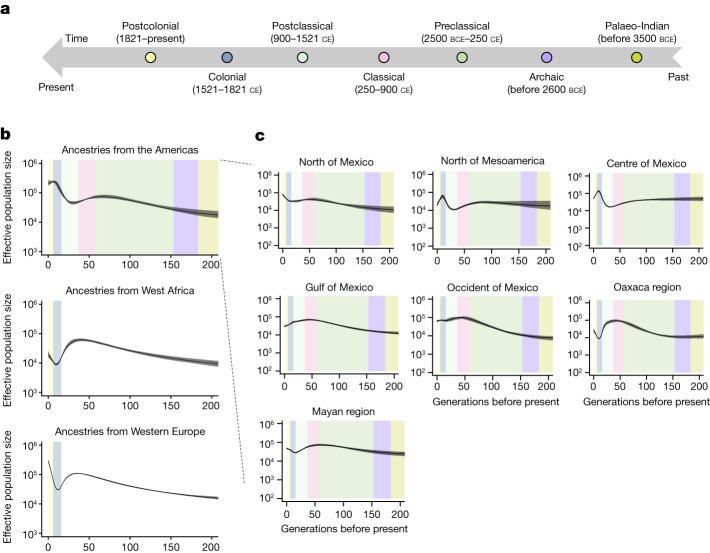
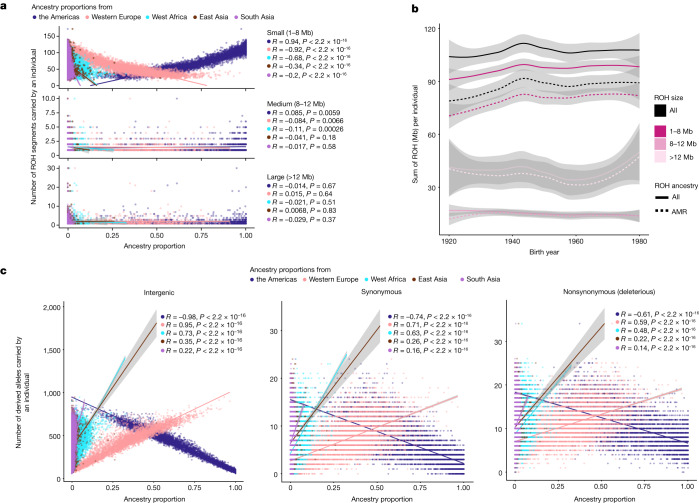
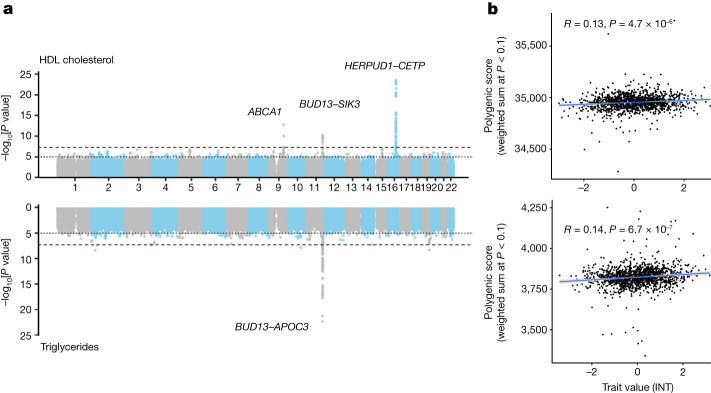
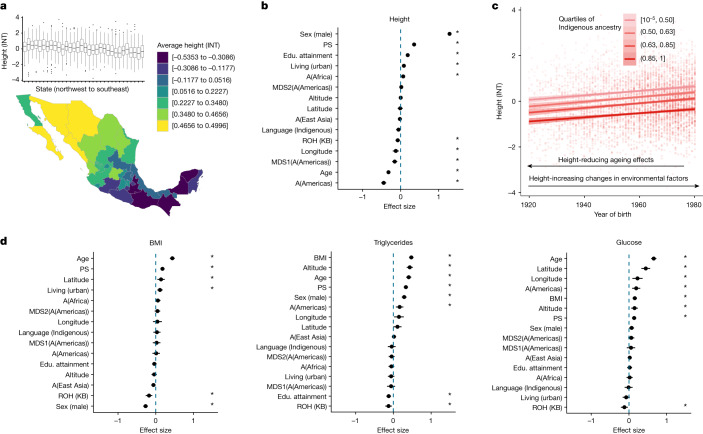
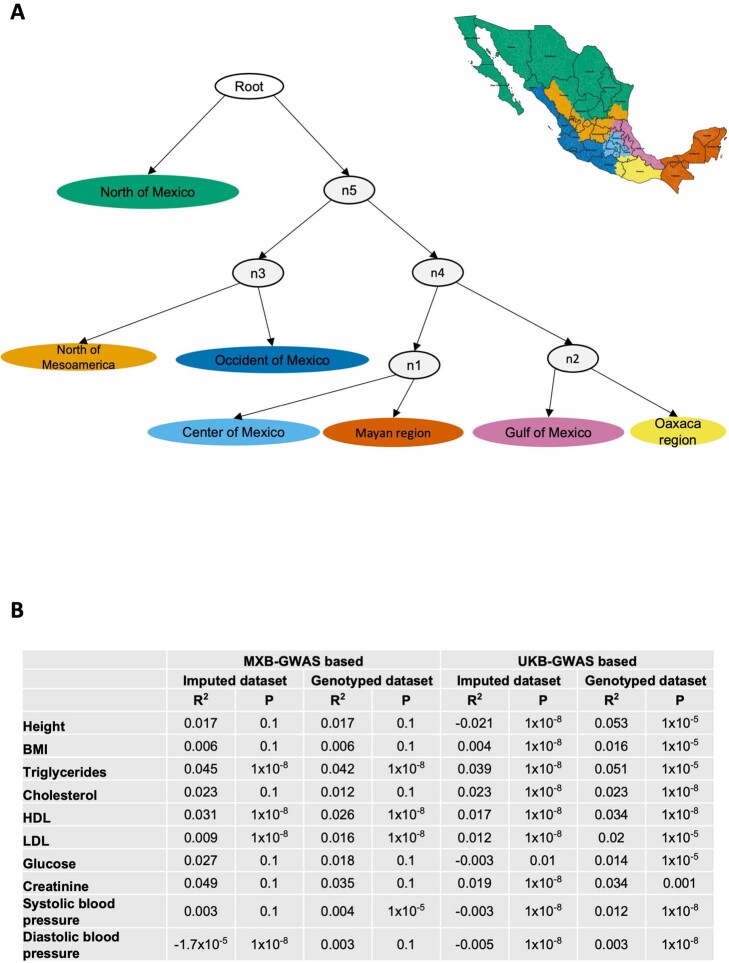
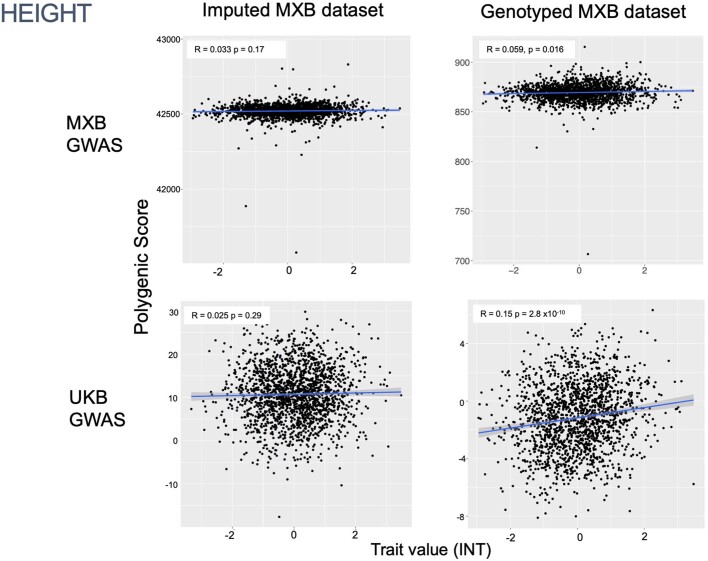
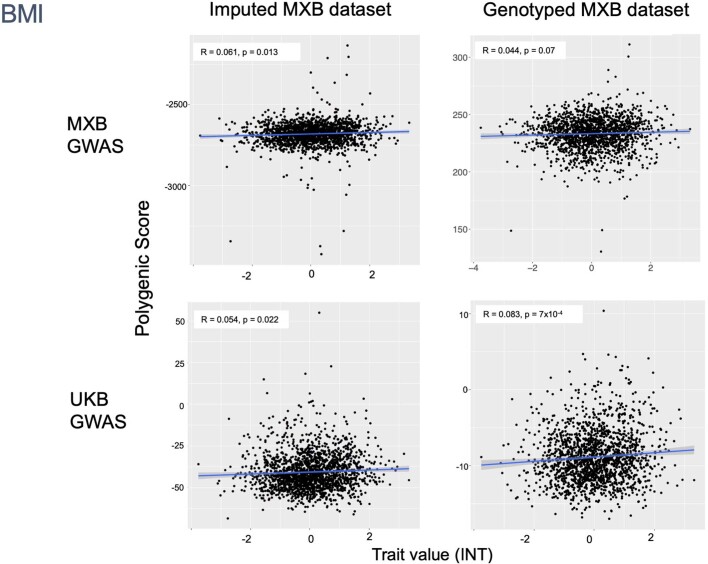
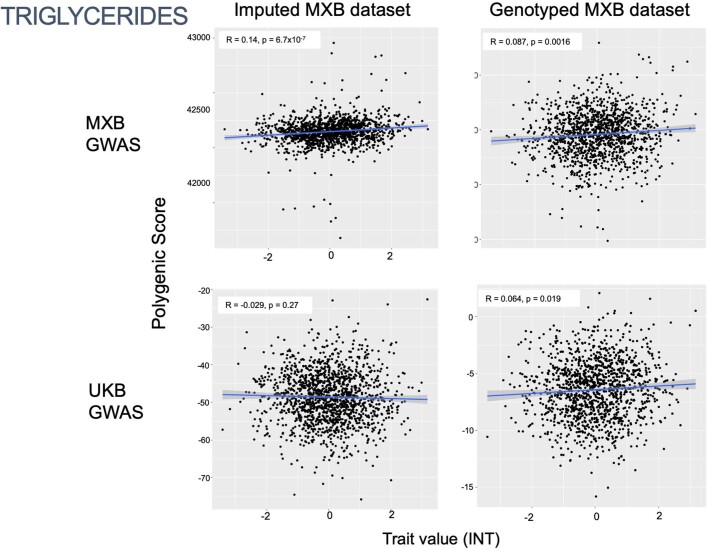
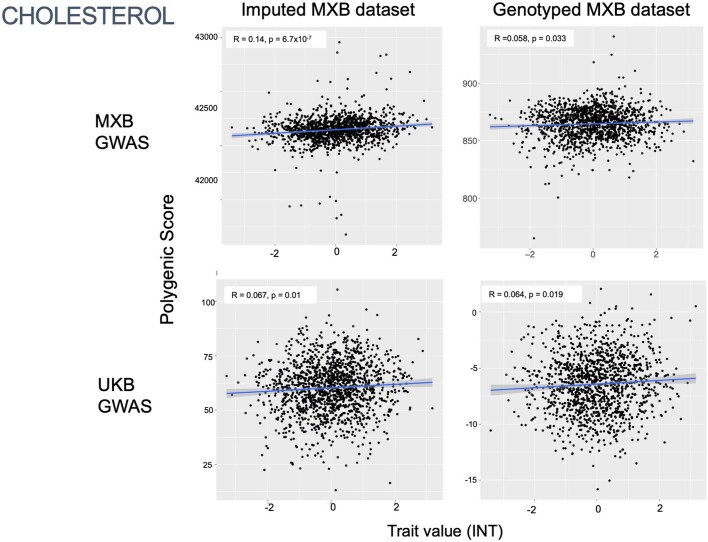
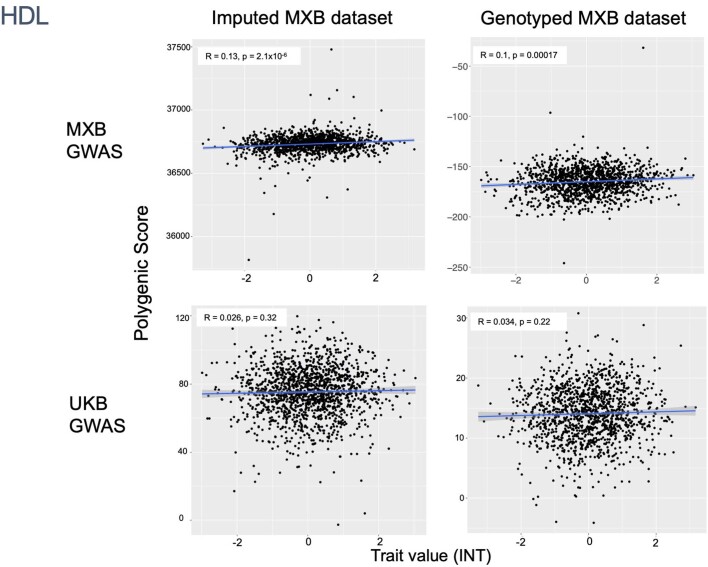
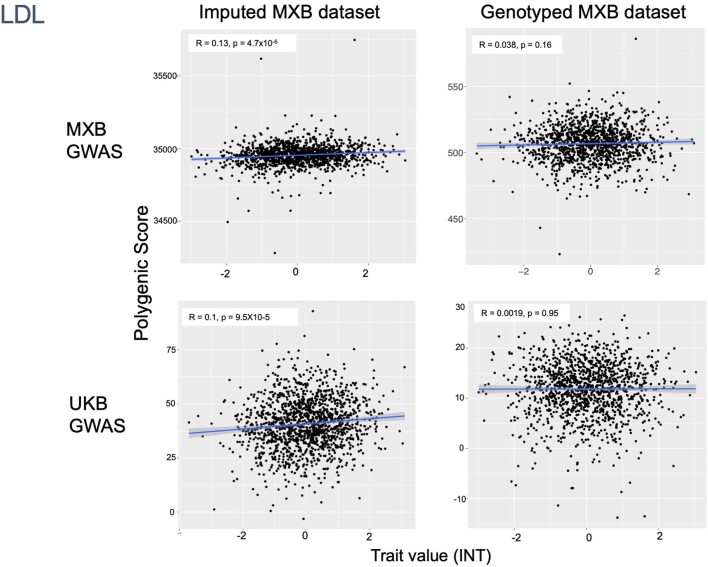
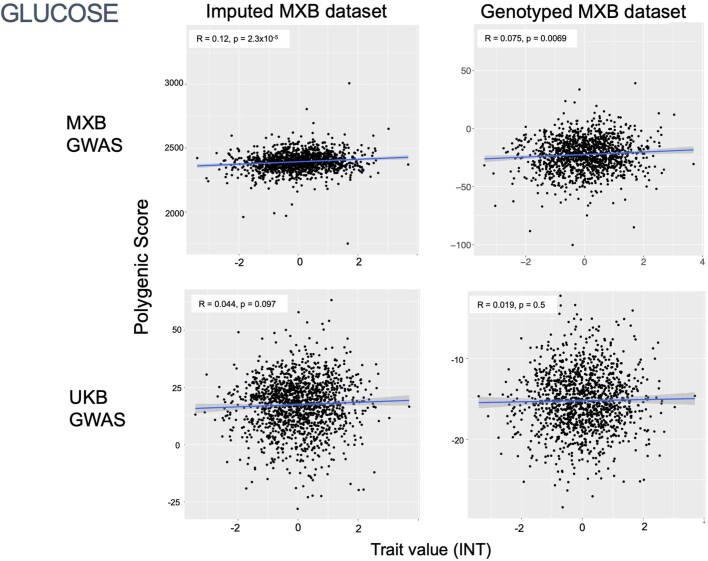
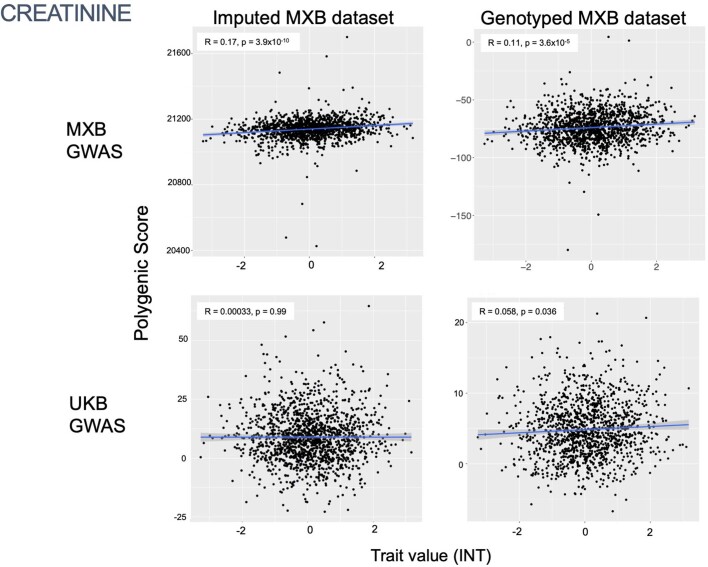
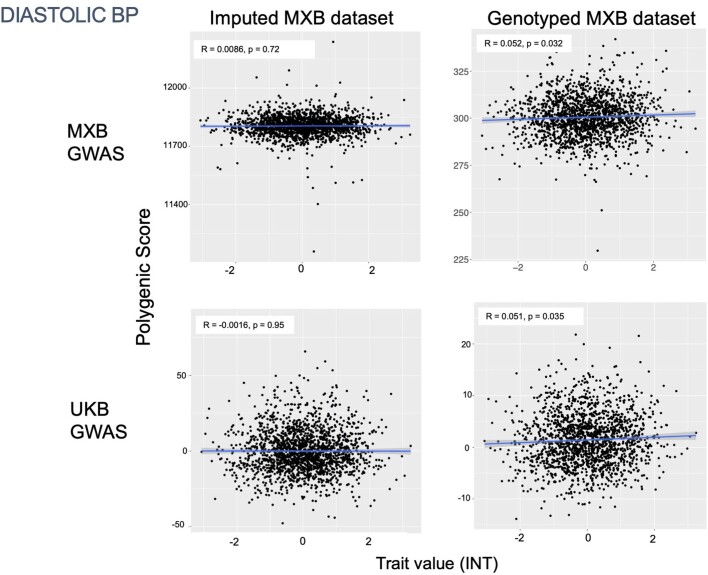
Latin America continues to be severely underrepresented in genomics research, and fine-scale genetic histories and complex trait architectures remain hidden owing to insufficient data1. To fill this gap, the Mexican Biobank project genotyped 6,057 individuals from 898 rural and urban localities across all 32 states in Mexico at a resolution of 1.8 million genome-wide markers with linked complex trait and disease information creating a valuable nationwide genotype-phenotype database. Here, using ancestry deconvolution and inference of identity-by-descent segments, we inferred ancestral population sizes across Mesoamerican regions over time, unravelling Indigenous, colonial and postcolonial demographic dynamics2-6. We observed variation in runs of homozygosity among genomic regions with different ancestries reflecting distinct demographic histories and, in turn, different distributions of rare deleterious variants. We conducted genome-wide association studies (GWAS) for 22 complex traits and found that several traits are better predicted using the Mexican Biobank GWAS compared to the UK Biobank GWAS7,8. We identified genetic and environmental factors associating with trait variation, such as the length of the genome in runs of homozygosity as a predictor for body mass index, triglycerides, glucose and height. This study provides insights into the genetic histories of individuals in Mexico and dissects their complex trait architectures, both crucial for making precision and preventive medicine initiatives accessible worldwide.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Hilmarsson, H., Kumar, A. S., Rastogi, R. & Bustamante, C. D. High resolution ancestry deconvolution for next generation genomic data. Preprint at bioRxiv10.1101/2021.09.19.460980 (2021).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
