

Genotyping, sequencing and analysis of 140,000 adults from Mexico City
- PMID: 37821707
- PMCID: PMC10600010
- DOI: 10.1038/s41586-023-06595-3
Genotyping, sequencing and analysis of 140,000 adults from Mexico City
Erratum in
-
Author Correction: Genotyping, sequencing and analysis of 140,000 adults from Mexico City.Nature. 2024 Feb;626(8001):E18. doi: 10.1038/s41586-024-07051-6. Nature. 2024. PMID: 38332034 Free PMC article. No abstract available.
Abstract
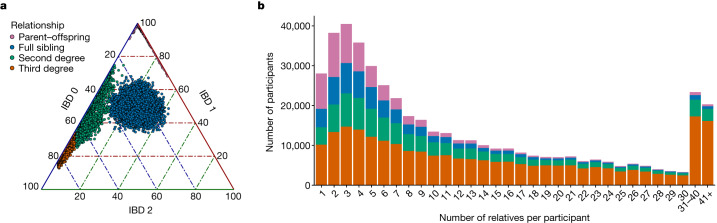
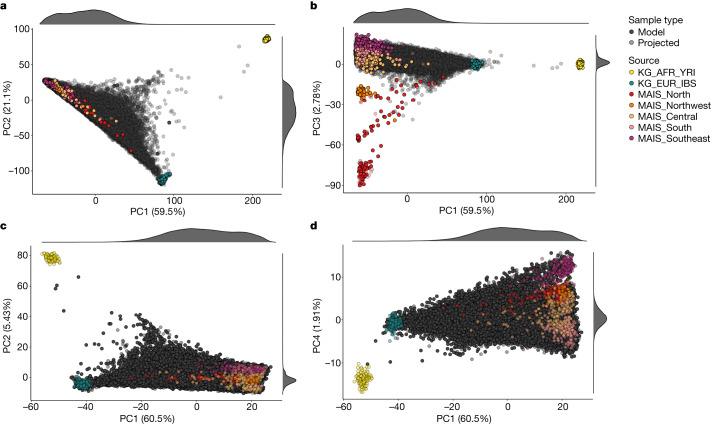
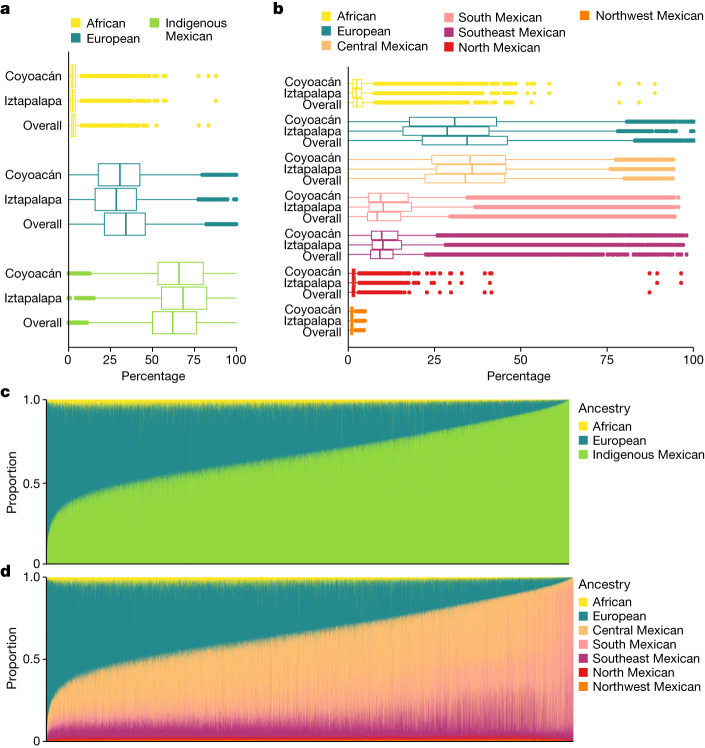
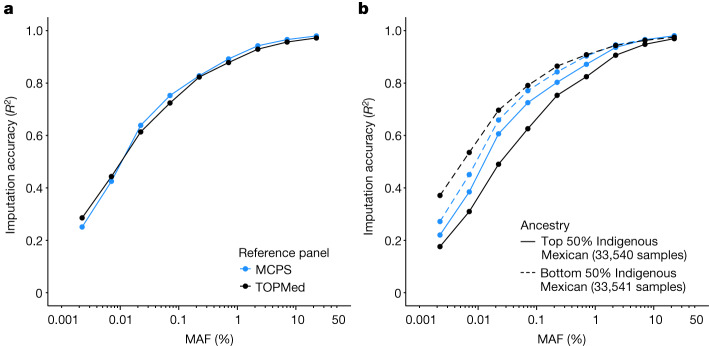
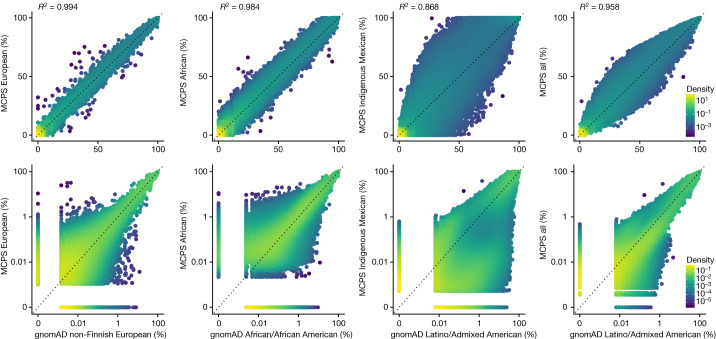
The Mexico City Prospective Study is a prospective cohort of more than 150,000 adults recruited two decades ago from the urban districts of Coyoacán and Iztapalapa in Mexico City1. Here we generated genotype and exome-sequencing data for all individuals and whole-genome sequencing data for 9,950 selected individuals. We describe high levels of relatedness and substantial heterogeneity in ancestry composition across individuals. Most sequenced individuals had admixed Indigenous American, European and African ancestry, with extensive admixture from Indigenous populations in central, southern and southeastern Mexico. Indigenous Mexican segments of the genome had lower levels of coding variation but an excess of homozygous loss-of-function variants compared with segments of African and European origin. We estimated ancestry-specific allele frequencies at 142 million genomic variants, with an effective sample size of 91,856 for Indigenous Mexican ancestry at exome variants, all available through a public browser. Using whole-genome sequencing, we developed an imputation reference panel that outperforms existing panels at common variants in individuals with high proportions of central, southern and southeastern Indigenous Mexican ancestry. Our work illustrates the value of genetic studies in diverse populations and provides foundational imputation and allele frequency resources for future genetic studies in Mexico and in the United States, where the Hispanic/Latino population is predominantly of Mexican descent.
© 2023. The Author(s).
Conflict of interest statement
A.Z., J. Backman, J. Mbatchou, S.M.G., T.J., Y.Z., D.L., J.S., R.P., A.P., X.B., S.B., L.H., R.L., A.L., E.M., M.J., A.D., N.L., C.P., E.J., W.S., J.O., J.R., T.A.T., G.A., A.B. and J. Marchini are current employees and/or stockholders of Regeneron Genetics Center or Regeneron Pharmaceuticals. A.N., K.R.S. and S.P. are current employees and/or stockholders of AstraZeneca. M.P. is a current employee and stockholder of AbbVie. All remaining authors declare no competing interests relevant to the current paper.
Figures










References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
