

A loss of function variant in AGPAT3 underlies intellectual disability and retinitis pigmentosa (IDRP) syndrome
- PMID: 37821758
- PMCID: PMC10689475
- DOI: 10.1038/s41431-023-01475-w
A loss of function variant in AGPAT3 underlies intellectual disability and retinitis pigmentosa (IDRP) syndrome
Abstract
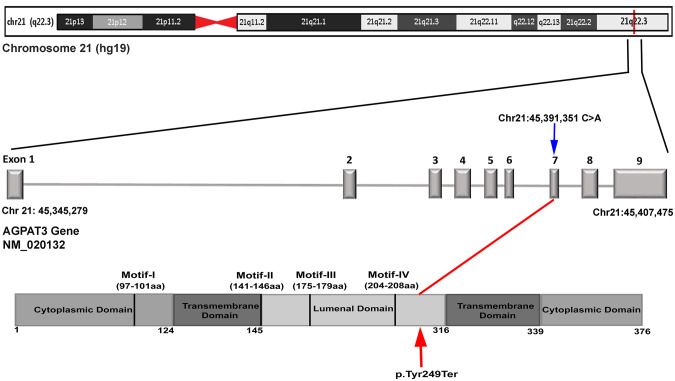
Intellectual disability (ID) and retinal dystrophy (RD) are the frequently found features of multiple syndromes involving additional systemic manifestations. Here, we studied a family with four members presenting severe ID and retinitis pigmentosa (RP). Using genome wide genotyping and exome sequencing, we identified a nonsense variant c.747 C > A (p.Tyr249Ter) in exon 7 of AGPAT3 which co-segregates with the disease phenotype. Western blot analysis of overexpressed WT and mutant AGPAT3 in HEK293T cells showed the absence of AGPAT3, suggesting instability of the truncated protein. Knockdown of Agpat3 in the embryonic mouse brain caused marked deficits in neuronal migration, strongly suggesting that reduced expression of AGPAT3 affects neuronal function. Altogether, our data indicates that AGPAT3 activity is essential for neuronal functioning and loss of its activity probably causes intellectual disability and retinitis pigmentosa (IDRP) syndrome.
© 2023. The Author(s), under exclusive licence to European Society of Human Genetics.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Usta M, Urganci N, Özçelik G, Çetinçelik Ü, Kafadar I, Özgüven BY. Joubert syndrome and related disorders: a rare cause of intrahepatic portal hypertension in childhood. Eur Rev Med Pharmacol Sci. 2015;19:2297–300. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
