

Endothelial lipid droplets suppress eNOS to link high fat consumption to blood pressure elevation
- PMID: 37824206
- PMCID: PMC10721151
- DOI: 10.1172/JCI173160
Endothelial lipid droplets suppress eNOS to link high fat consumption to blood pressure elevation
Abstract
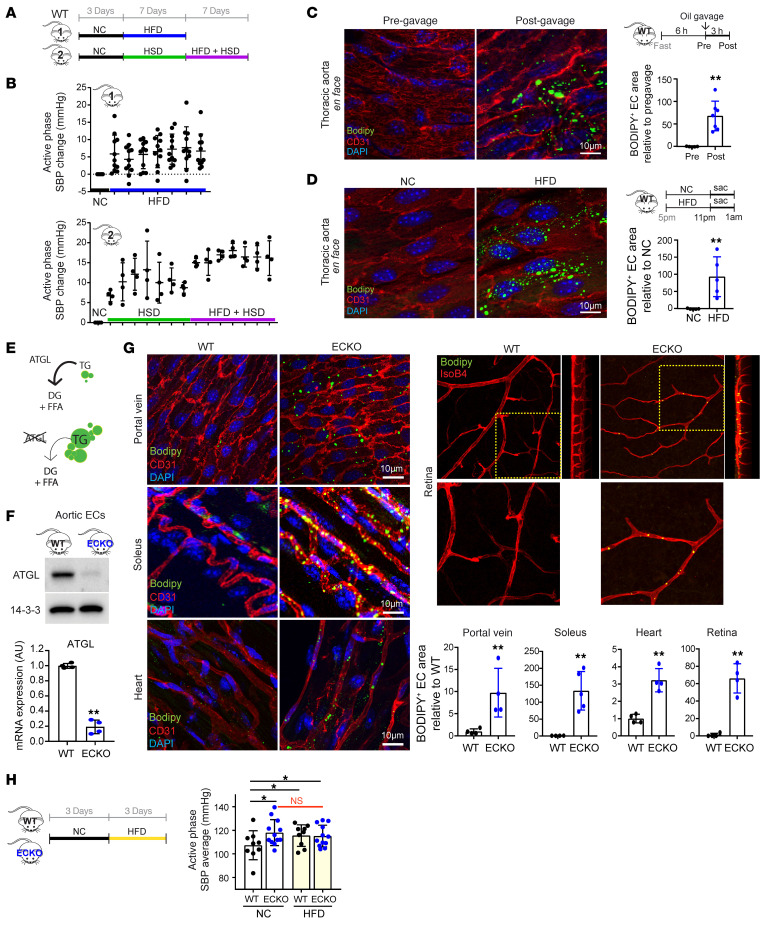
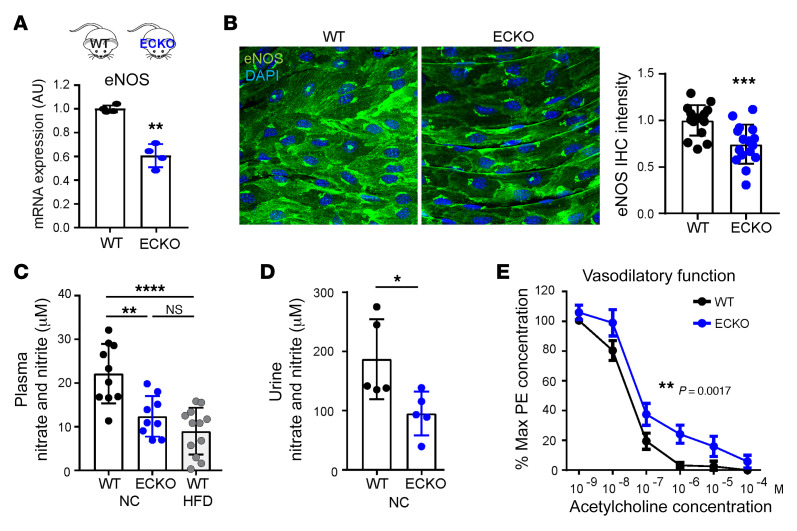
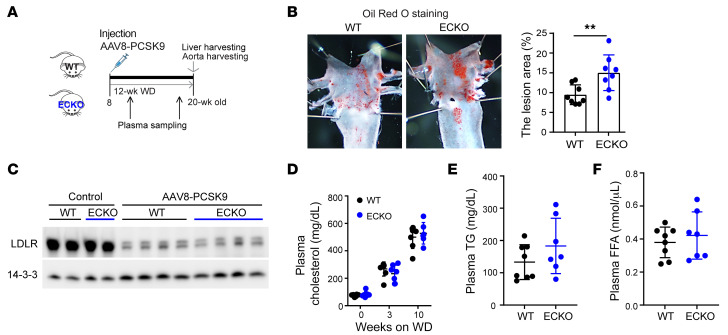
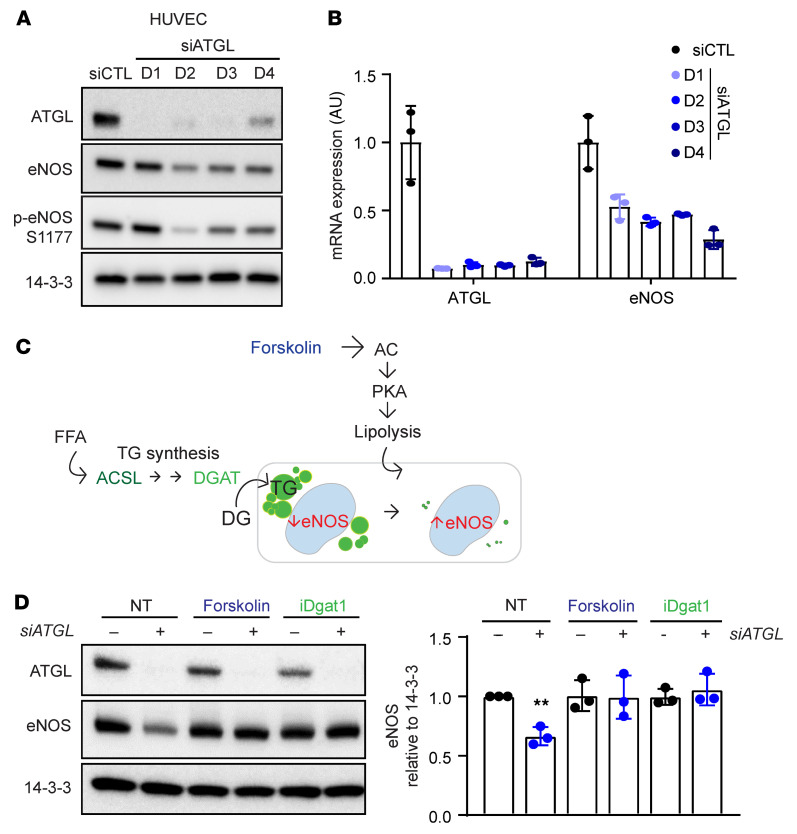
Metabolic syndrome, today affecting more than 20% of the US population, is a group of 5 conditions that often coexist and that strongly predispose to cardiovascular disease. How these conditions are linked mechanistically remains unclear, especially two of these: obesity and elevated blood pressure. Here, we show that high fat consumption in mice leads to the accumulation of lipid droplets in endothelial cells throughout the organism and that lipid droplet accumulation in endothelium suppresses endothelial nitric oxide synthase (eNOS), reduces NO production, elevates blood pressure, and accelerates atherosclerosis. Mechanistically, the accumulation of lipid droplets destabilizes eNOS mRNA and activates an endothelial inflammatory signaling cascade that suppresses eNOS and NO production. Pharmacological prevention of lipid droplet formation reverses the suppression of NO production in cell culture and in vivo and blunts blood pressure elevation in response to a high-fat diet. These results highlight lipid droplets as a critical and unappreciated component of endothelial cell biology, explain how lipids increase blood pressure acutely, and provide a mechanistic account for the epidemiological link between obesity and elevated blood pressure.
Keywords: Cardiology; Endothelial cells.
Conflict of interest statement
Figures
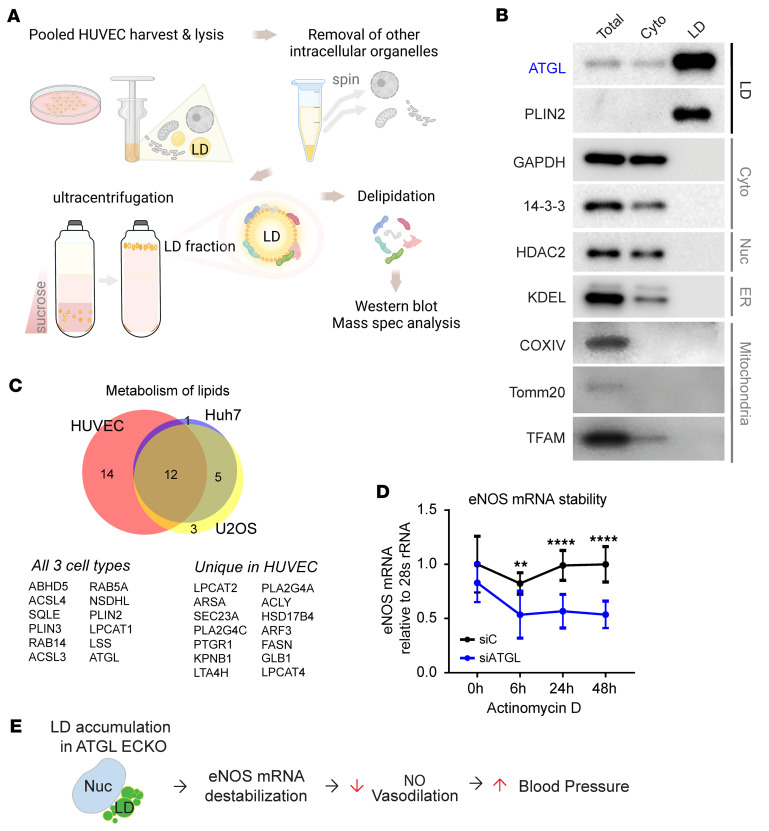
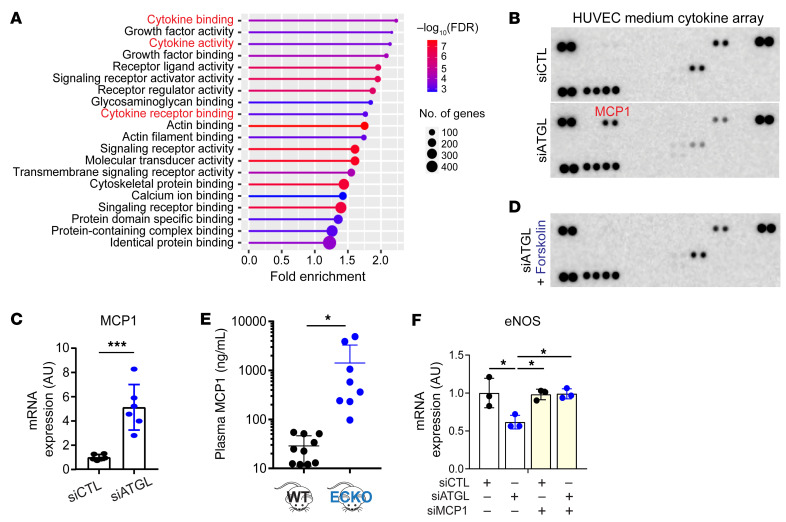
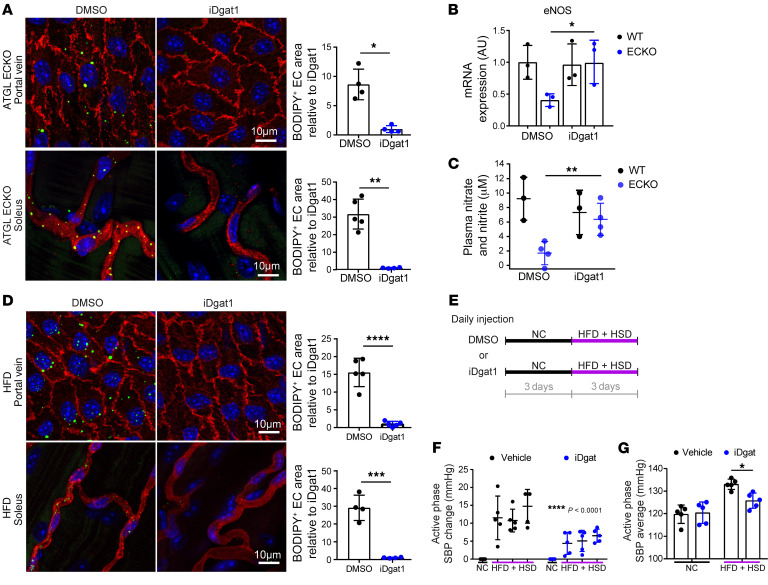
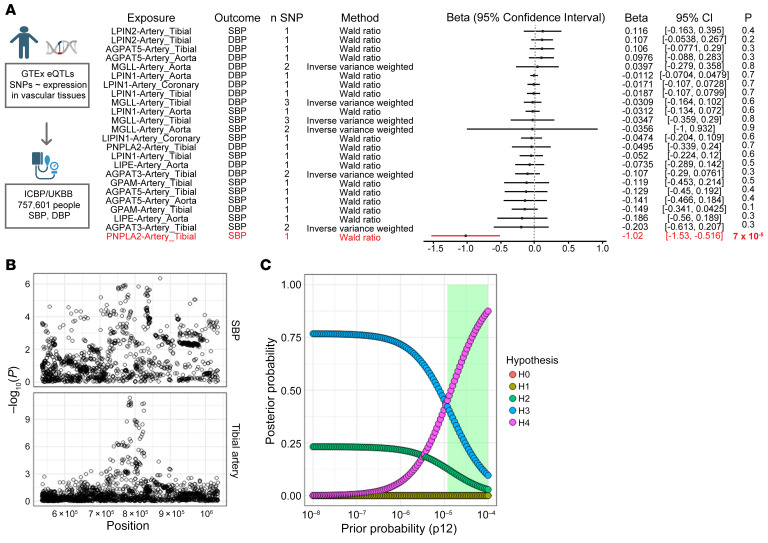
References
-
- Grekin RJ, et al. Mechanisms in the pressor effects of hepatic portal venous fatty acid infusion. Am J Physiol. 1997;273(1 pt 2):R324–R330. - PubMed
