

Assessing the performance of methods for cell clustering from single-cell DNA sequencing data
- PMID: 37824596
- PMCID: PMC10597505
- DOI: 10.1371/journal.pcbi.1010480
Assessing the performance of methods for cell clustering from single-cell DNA sequencing data
Abstract
Background: Many cancer genomes have been known to contain more than one subclone inside one tumor, the phenomenon of which is called intra-tumor heterogeneity (ITH). Characterizing ITH is essential in designing treatment plans, prognosis as well as the study of cancer progression. Single-cell DNA sequencing (scDNAseq) has been proven effective in deciphering ITH. Cells corresponding to each subclone are supposed to carry a unique set of mutations such as single nucleotide variations (SNV). While there have been many studies on the cancer evolutionary tree reconstruction, not many have been proposed that simply characterize the subclonality without tree reconstruction. While tree reconstruction is important in the study of cancer evolutionary history, typically they are computationally expensive in terms of running time and memory consumption due to the huge search space of the tree structure. On the other hand, subclonality characterization of single cells can be converted into a cell clustering problem, the dimension of which is much smaller, and the turnaround time is much shorter. Despite the existence of a few state-of-the-art cell clustering computational tools for scDNAseq, there lacks a comprehensive and objective comparison under different settings.
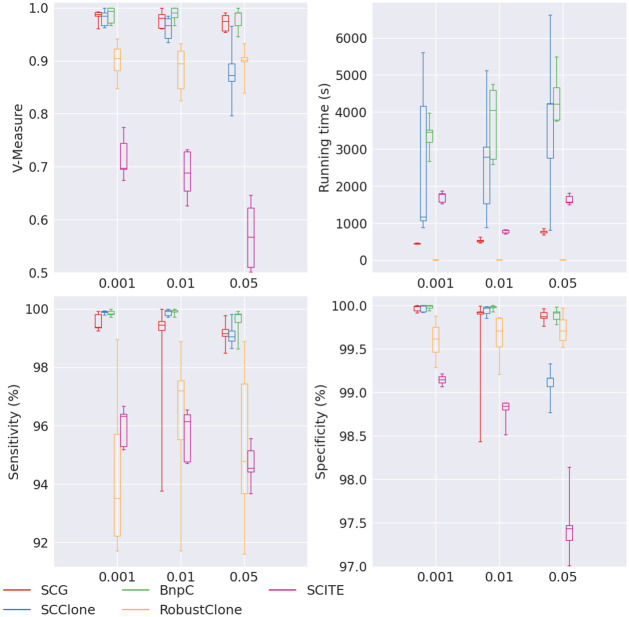
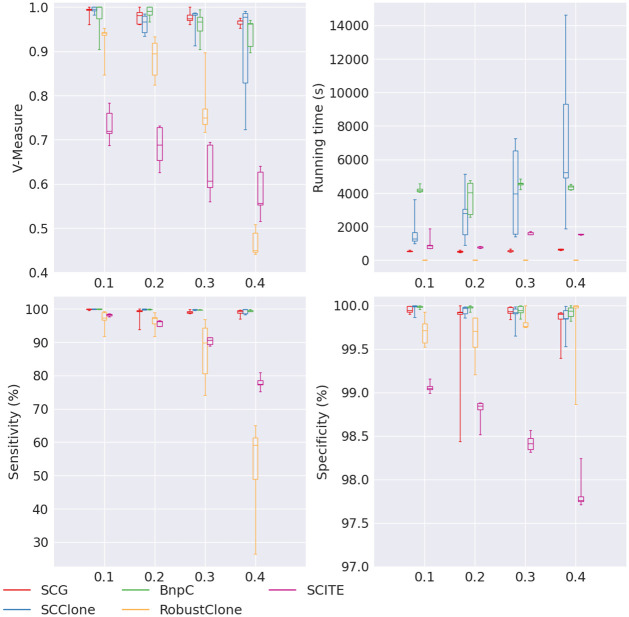
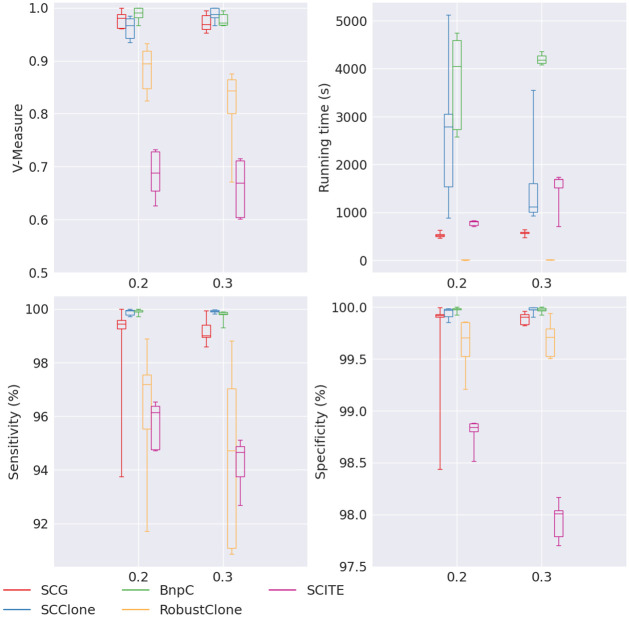
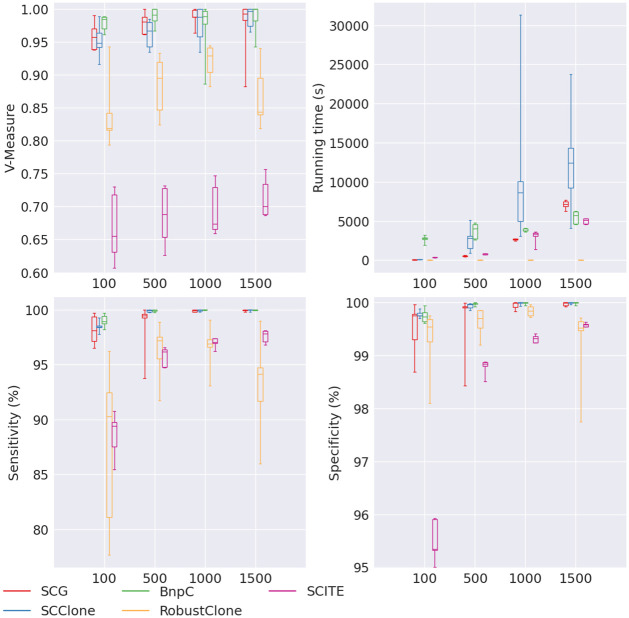
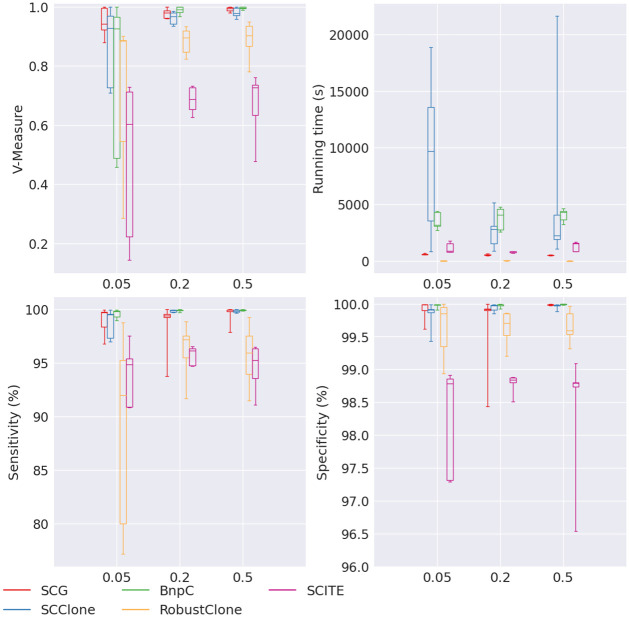
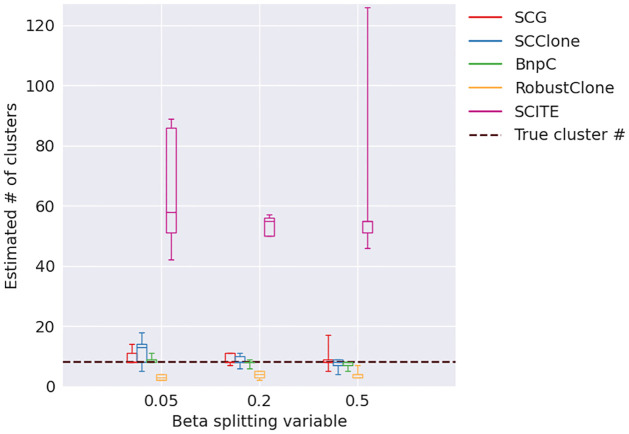
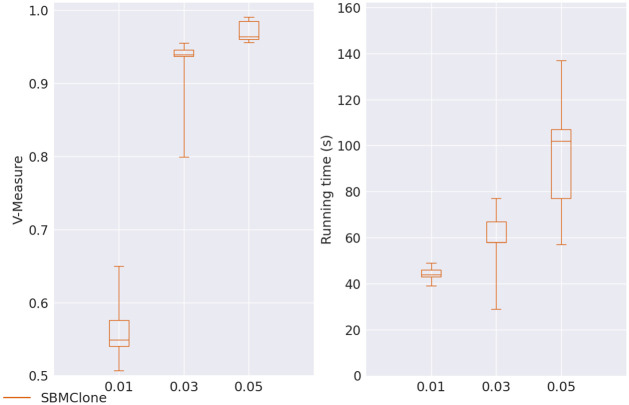
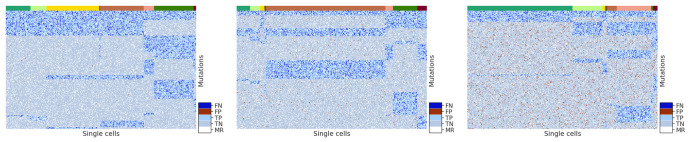
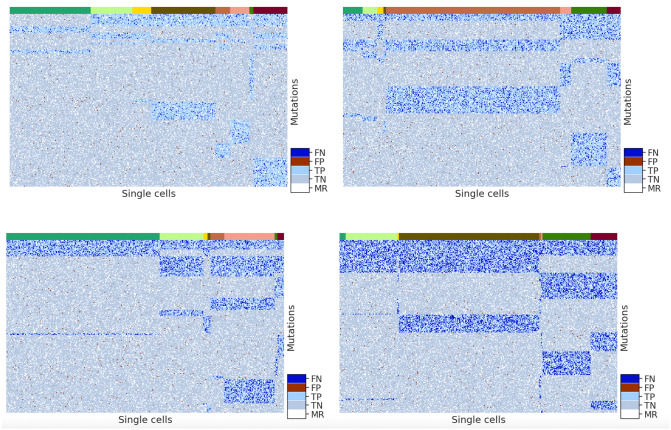
Results: In this paper, we evaluated six state-of-the-art cell clustering tools-SCG, BnpC, SCClone, RobustClone, SCITE and SBMClone-on simulated data sets given a variety of parameter settings and a real data set. We designed a simulator specifically for cell clustering, and compared these methods' performances in terms of their clustering accuracy, specificity and sensitivity and running time. For SBMClone, we specifically designed an ultra-low coverage large data set to evaluate its performance in the face of an extremely high missing rate.
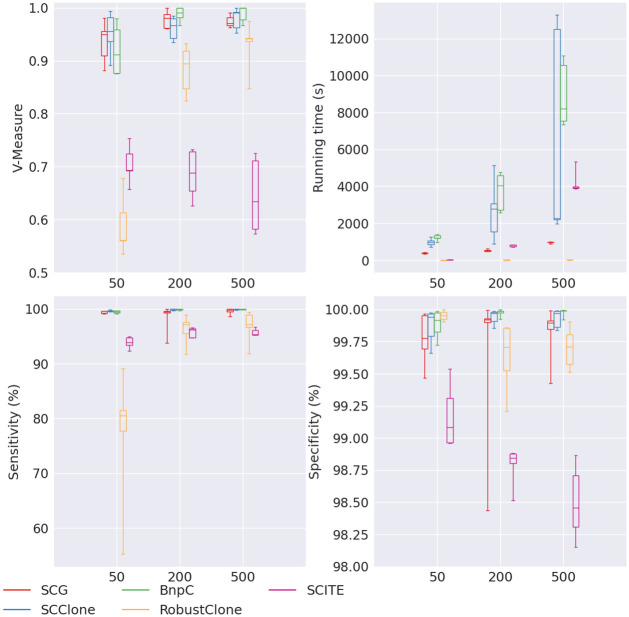
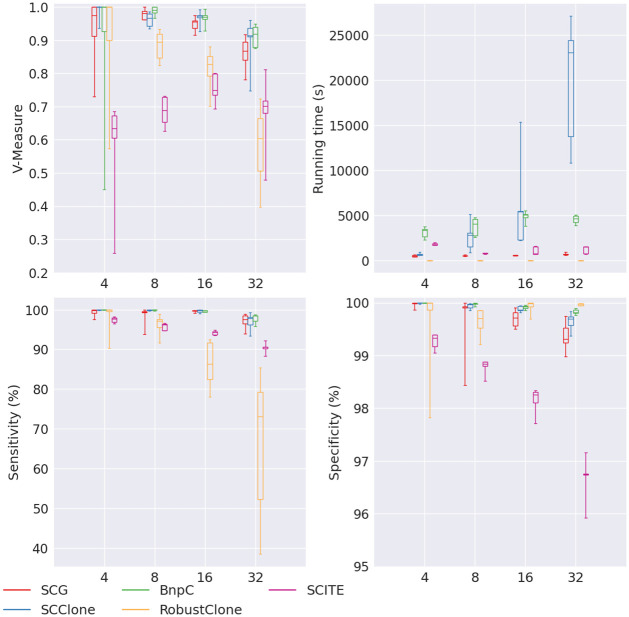
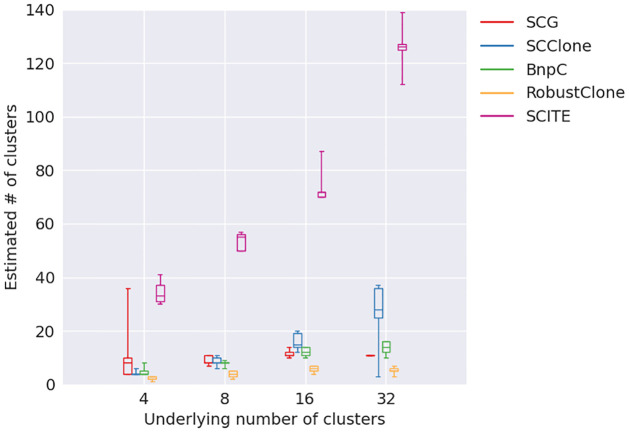
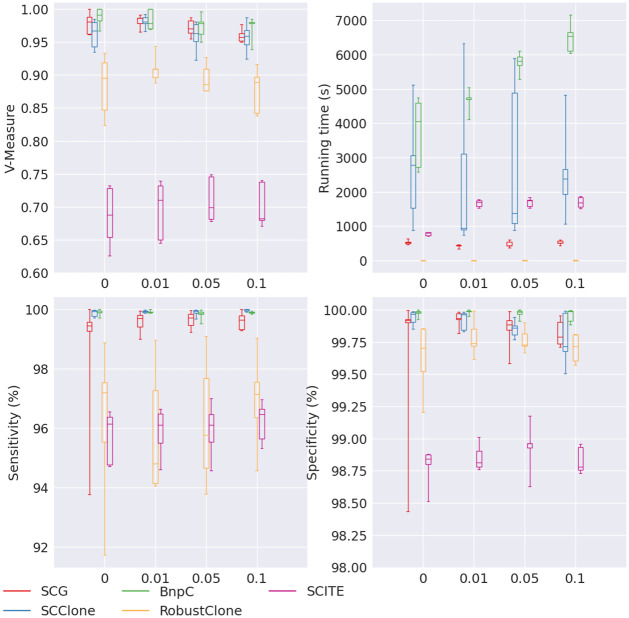
Conclusion: From the benchmark study, we conclude that BnpC and SCG's clustering accuracy are the highest and comparable to each other. However, BnpC is more advantageous in terms of running time when cell number is high (> 1500). It also has a higher clustering accuracy than SCG when cluster number is high (> 16). SCClone's accuracy in estimating the number of clusters is the highest. RobustClone and SCITE's clustering accuracy are the lowest for all experiments. SCITE tends to over-estimate the cluster number and has a low specificity, whereas RobustClone tends to under-estimate the cluster number and has a much lower sensitivity than other methods. SBMClone produced reasonably good clustering (V-measure > 0.9) when coverage is > = 0.03 and thus is highly recommended for ultra-low coverage large scDNAseq data sets.
Copyright: © 2023 Khan, Mallory. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
