

Multi-omic profiling of the developing human cerebral cortex at the single-cell level
- PMID: 37824614
- PMCID: PMC10569714
- DOI: 10.1126/sciadv.adg3754
Multi-omic profiling of the developing human cerebral cortex at the single-cell level
Abstract
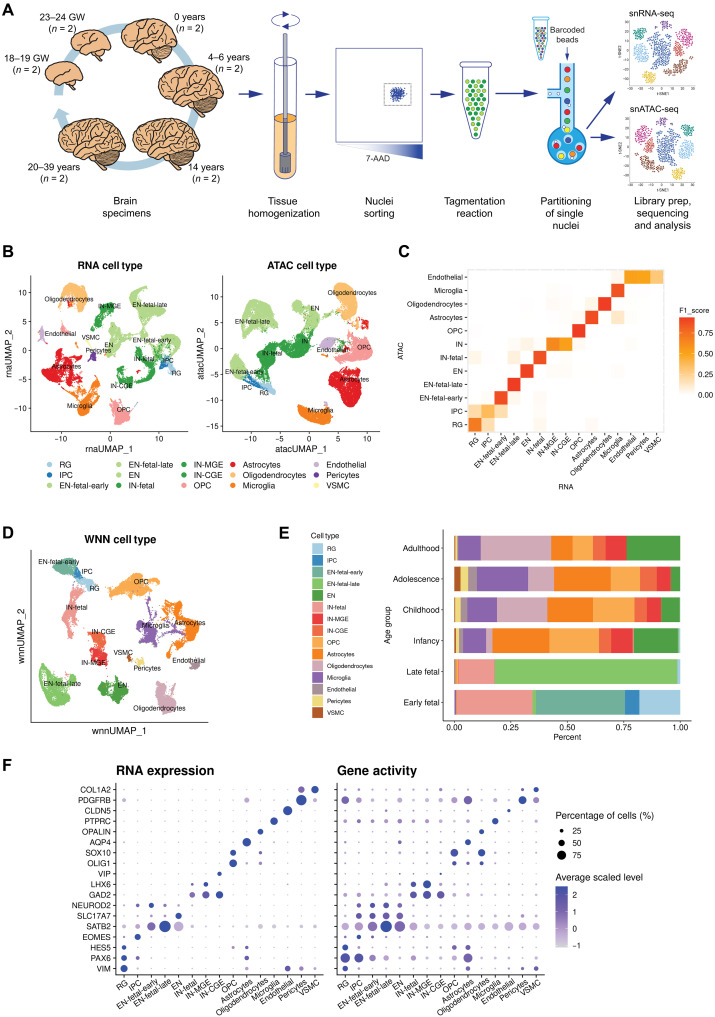
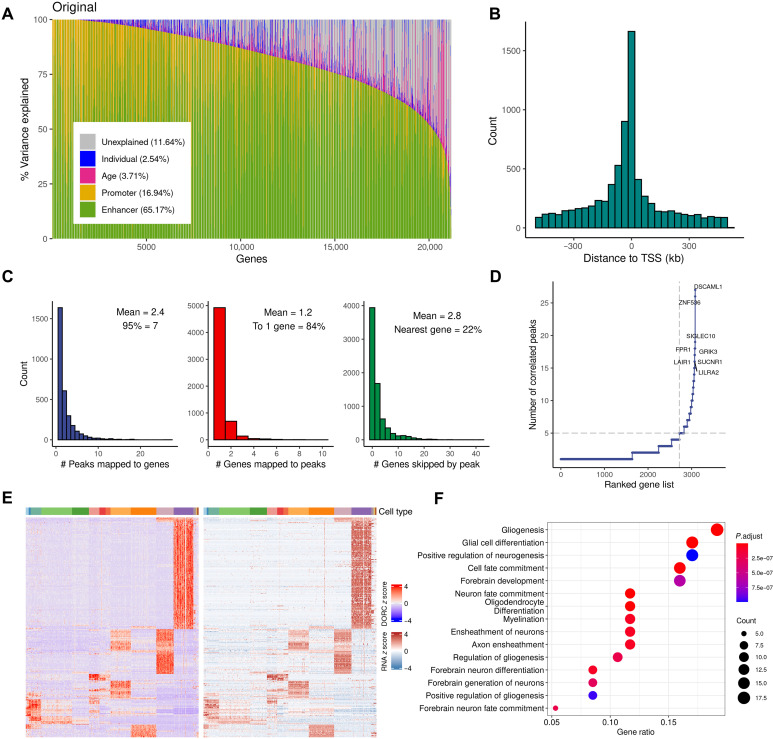
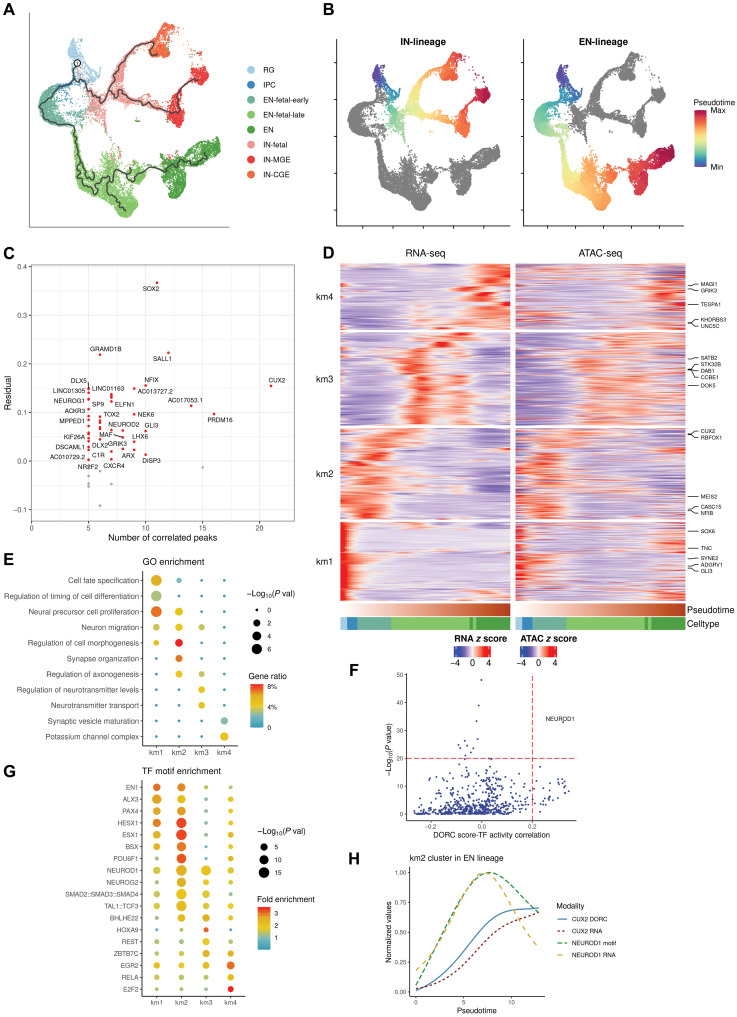
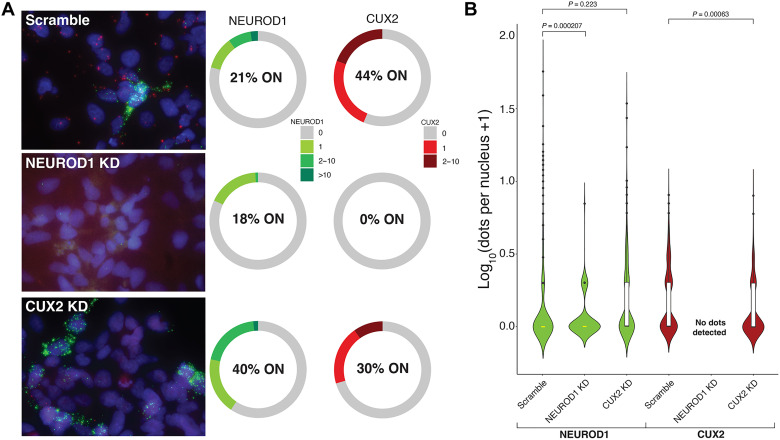
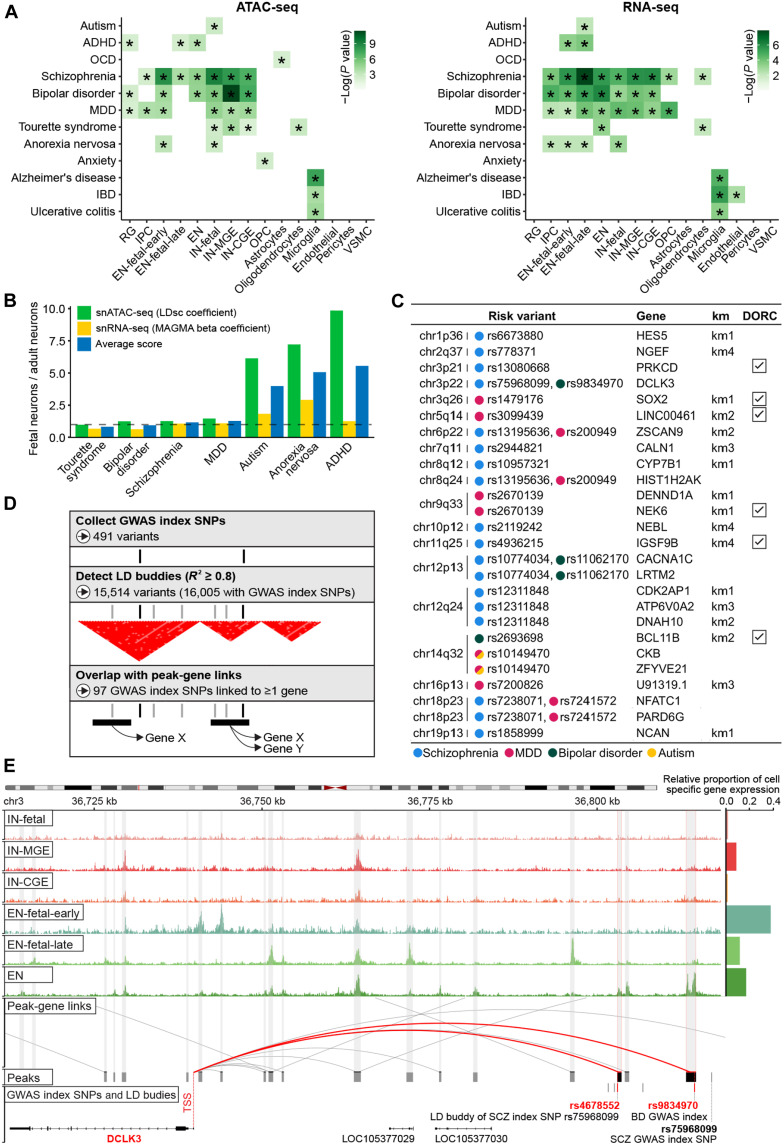
The cellular complexity of the human brain is established via dynamic changes in gene expression throughout development that is mediated, in part, by the spatiotemporal activity of cis-regulatory elements (CREs). We simultaneously profiled gene expression and chromatin accessibility in 45,549 cortical nuclei across six broad developmental time points from fetus to adult. We identified cell type-specific domains in which chromatin accessibility is highly correlated with gene expression. Differentiation pseudotime trajectory analysis indicates that chromatin accessibility at CREs precedes transcription and that dynamic changes in chromatin structure play a critical role in neuronal lineage commitment. In addition, we mapped cell type-specific and temporally specific genetic loci implicated in neuropsychiatric traits, including schizophrenia and bipolar disorder. Together, our results describe the complex regulation of cell composition at critical stages in lineage determination and shed light on the impact of spatiotemporal alterations in gene expression on neuropsychiatric disease.
Figures
References
-
- Birnbaum R., Weinberger D. R., Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 18, 727–740 (2017). - PubMed
-
- Kang H. J., Kawasawa Y. I., Cheng F., Zhu Y., Xu X., Li M., Sousa A. M. M., Pletikos M., Meyer K. A., Sedmak G., Guennel T., Shin Y., Johnson M. B., Krsnik Ž., Mayer S., Fertuzinhos S., Umlauf S., Lisgo S. N., Vortmeyer A., Weinberger D. R., Mane S., Hyde T. M., Huttner A., Reimers M., Kleinman J. E., Šestan N., Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
