

Ibrutinib-based therapy reinvigorates CD8+ T cells compared to chemoimmunotherapy: immune monitoring from the E1912 trial
- PMID: 37824808
- PMCID: PMC10797553
- DOI: 10.1182/blood.2023020554
Ibrutinib-based therapy reinvigorates CD8+ T cells compared to chemoimmunotherapy: immune monitoring from the E1912 trial
Abstract
Bruton tyrosine kinase inhibitors (BTKis) that target B-cell receptor signaling have led to a paradigm shift in chronic lymphocytic leukemia (CLL) treatment. BTKis have been shown to reduce abnormally high CLL-associated T-cell counts and the expression of immune checkpoint receptors concomitantly with tumor reduction. However, the impact of BTKi therapy on T-cell function has not been fully characterized. Here, we performed longitudinal immunophenotypic and functional analysis of pretreatment and on-treatment (6 and 12 months) peripheral blood samples from patients in the phase 3 E1912 trial comparing ibrutinib-rituximab with fludarabine, cyclophosphamide, and rituximab (FCR). Intriguingly, we report that despite reduced overall T-cell counts; higher numbers of T cells, including effector CD8+ subsets at baseline and at the 6-month time point, associated with no infections; and favorable progression-free survival in the ibrutinib-rituximab arm. Assays demonstrated enhanced anti-CLL T-cell killing function during ibrutinib-rituximab treatment, including a switch from predominantly CD4+ T-cell:CLL immune synapses at baseline to increased CD8+ lytic synapses on-therapy. Conversely, in the FCR arm, higher T-cell numbers correlated with adverse clinical responses and showed no functional improvement. We further demonstrate the potential of exploiting rejuvenated T-cell cytotoxicity during ibrutinib-rituximab treatment, using the bispecific antibody glofitamab, supporting combination immunotherapy approaches.
© 2024 by American Society of Hematology. Published by Elsevier Inc. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: S.H., M.B., and C.K. are employees of and declare patents and stock ownership with Roche Glycart AG. A.G.R. has received research support from Roche Glycart AG to Institution as part of a research collaboration. The remaining authors declare no competing financial interests.
Figures
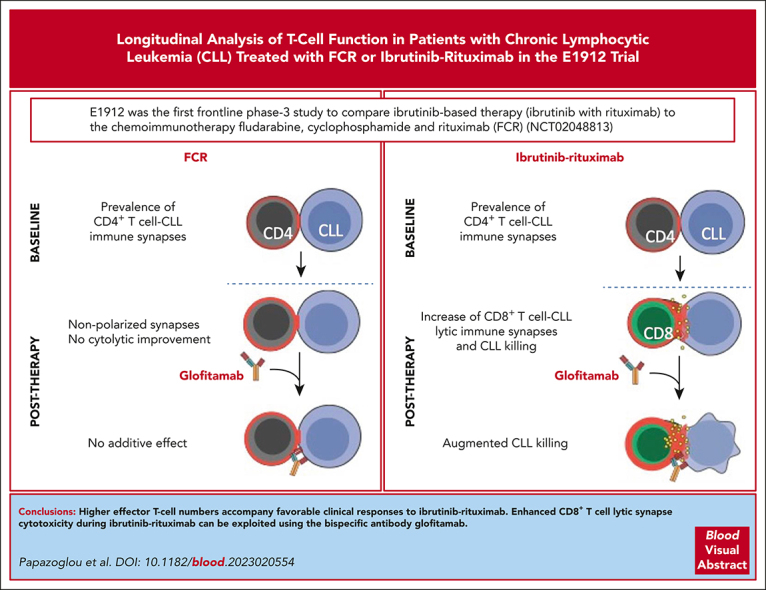
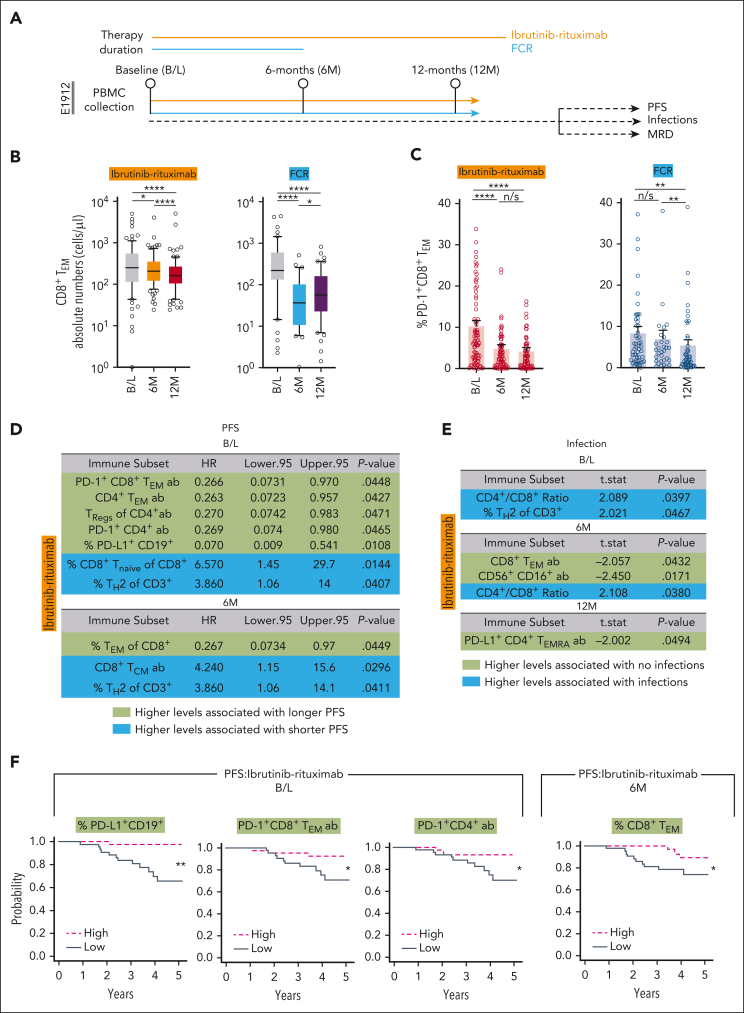
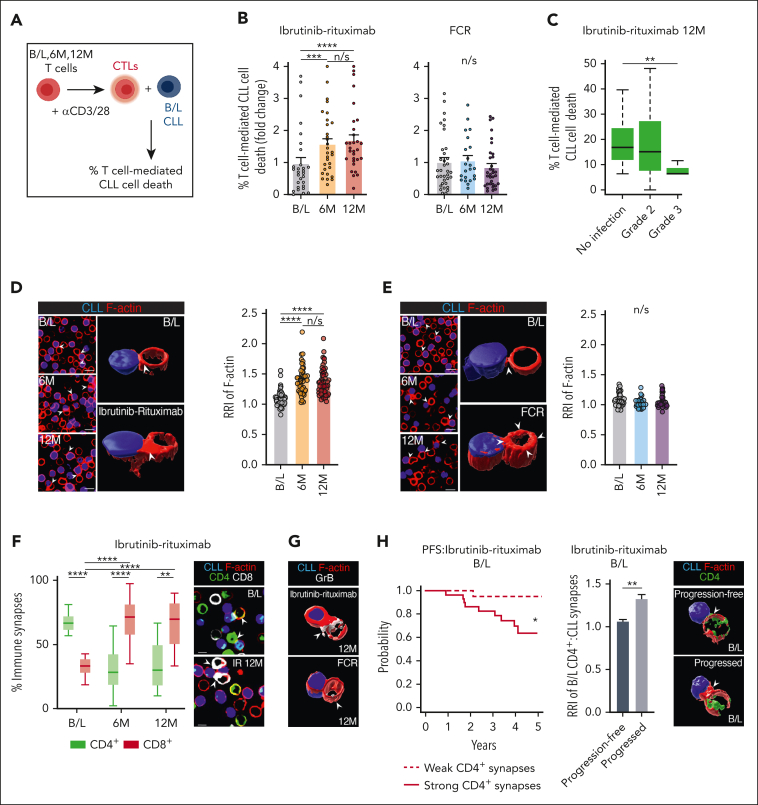
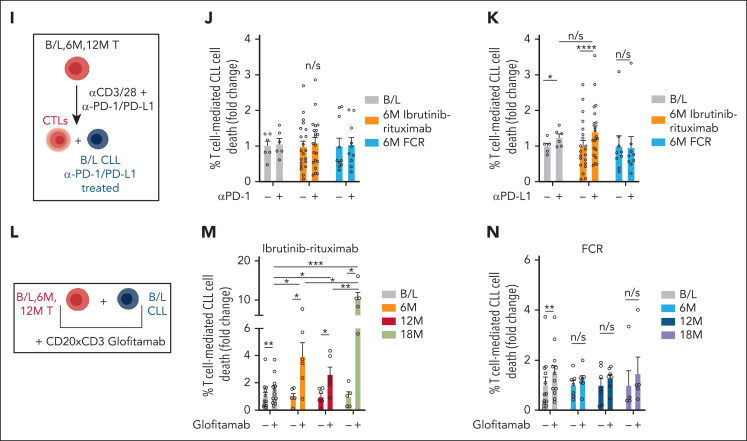
Comment in
-
Ibrutinib reversal of immune exhaustion in CLL.Blood. 2024 Jan 4;143(1):5-7. doi: 10.1182/blood.2023022243. Blood. 2024. PMID: 38175677 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
