

A Ubiquitin-Dependent Switch on MEF2D Senses Pro-Metastatic Niche Signals to Facilitate Intrahepatic Metastasis of Liver Cancer
- PMID: 37828611
- PMCID: PMC10724427
- DOI: 10.1002/advs.202305550
A Ubiquitin-Dependent Switch on MEF2D Senses Pro-Metastatic Niche Signals to Facilitate Intrahepatic Metastasis of Liver Cancer
Abstract
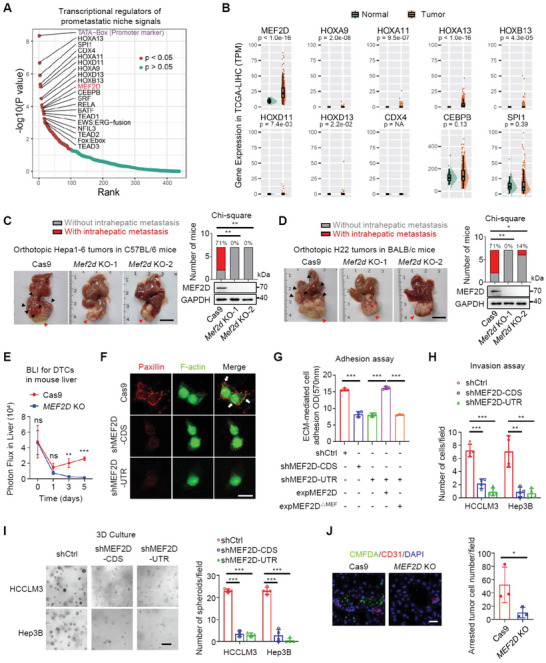
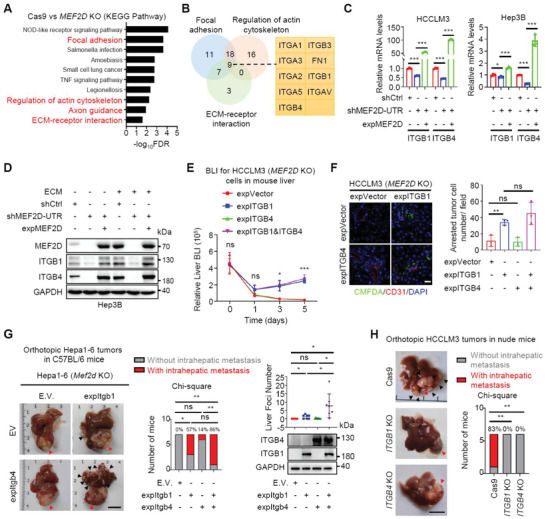
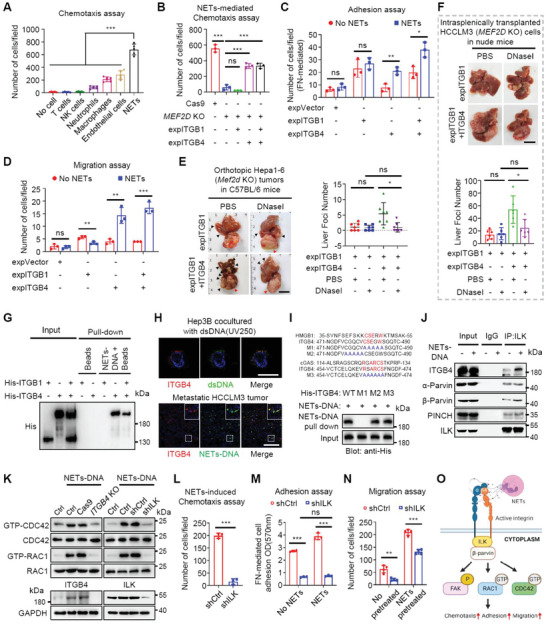
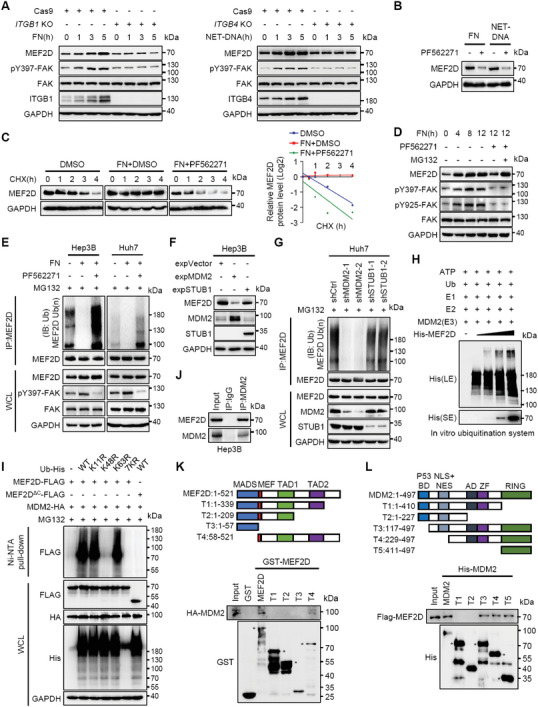
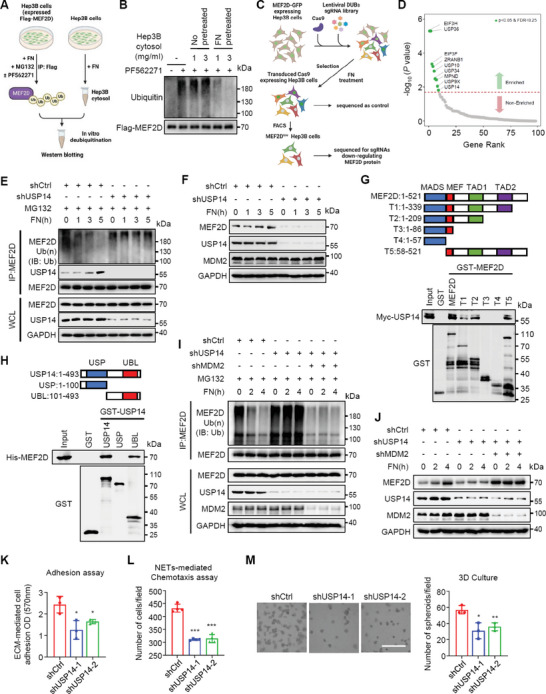
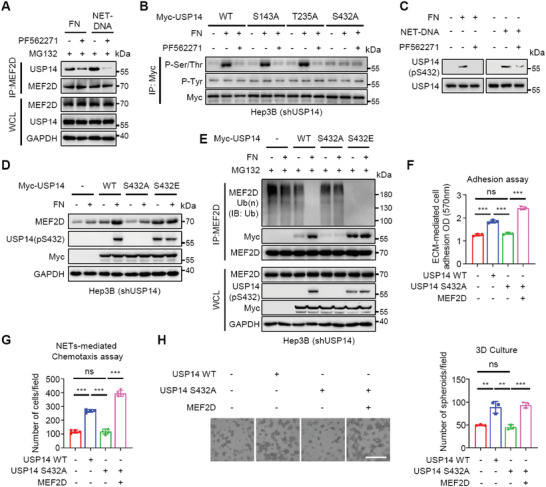
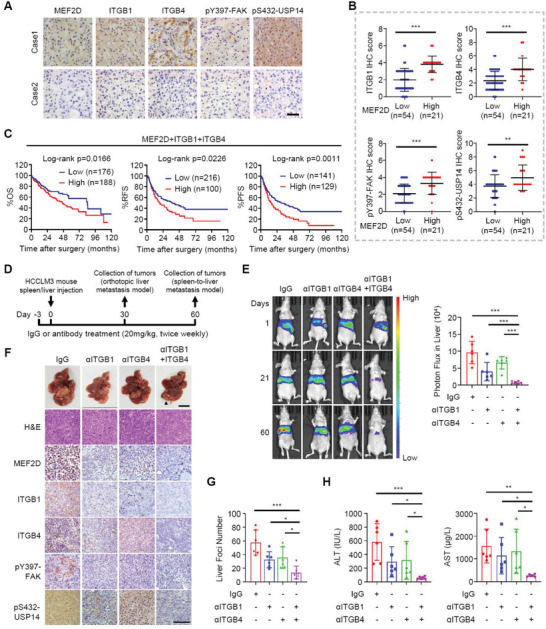
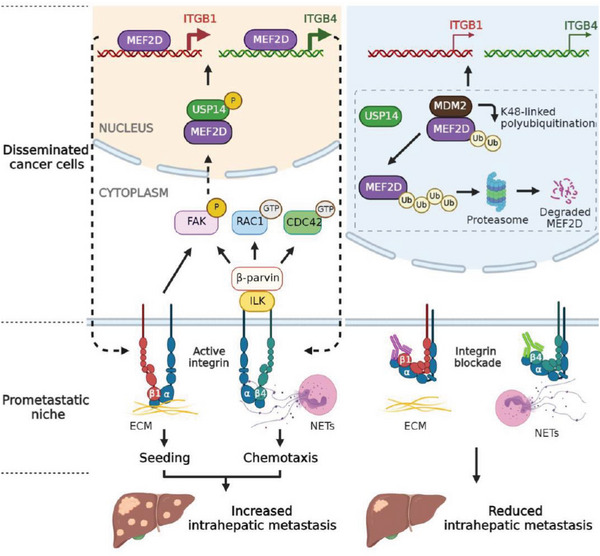
Effective treatment for metastasis, a leading cause of cancer-associated death, is still lacking. To seed on a distal organ, disseminated cancer cells (DCCs) must adapt to the local tissue microenvironment. However, it remains elusive how DCCs respond the pro-metastatic niche signals. Here, systemic motif-enrichment identified myocyte enhancer factor 2D (MEF2D) as a critical sensor of niche signals to regulate DCCs adhesion and colonization, leading to intrahepatic metastasis and recurrence of liver cancer. In this context, MEF2D transactivates Itgb1 (coding β1-integrin) and Itgb4 (coding β4-integrin) to execute temporally unique functions, where ITGB1 recognizes extracellular matrix for early seeding, and ITGB4 acts as a novel sensor of neutrophil extracellular traps-DNA (NETs-DNA) for subsequent chemotaxis and colonization. In turn, an integrin-FAK circuit promotes a phosphorylation-dependent USP14-orchastrated deubiquitination switch to stabilize MEF2D via circumventing degradation by the E3-ubiquitin-ligase MDM2. Clinically, the USP14(pS432)-MEF2D-ITGB1/4 feedback loop is often hyper-active and indicative of inferior outcomes in human malignancies, while its blockade abrogated intrahepatic metastasis of DCCs. Together, DCCs exploit a deubiquitination-dependent switch on MEF2D to integrate niche signals in the liver mesenchyme, thereby amplifying the pro-metastatic integrin-FAK signaling. Disruption of this feedback loop is clinically applicable with fast-track potential to block microenvironmental cues driving metastasis.
Keywords: MEF2D; disseminated cancer cells; integrin; neutrophil extracellular traps; pro-metastatic niche.
© 2023 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Celià‐Terrassa T., Kang Y., Nat. Cell Biol. 2018, 20, 868. - PubMed
-
- Fane M. E., Chhabra Y., Alicea G. M., Maranto D. A., Douglass S. M., Webster M. R., Rebecca V. W., Marino G. E., Almeida F., Ecker B. L., Zabransky D. J., Hüser L., Beer T., Tang H.‐Y., Kossenkov A., Herlyn M., Speicher D. W., Xu W., Xu X., Jaffee E. M., Aguirre‐Ghiso J. A., Weeraratna A. T., Nature 2022, 606, 396. - PMC - PubMed
-
- Lee J. W., Stone M. L., Porrett P. M., Thomas S. K., Komar C. A., Li J. H., Delman D., Graham K., Gladney W. L., Hua X., Black T. A., Chien A. L., Majmundar K. S., Thompson J. C., Yee S. S., O'Hara M. H., Aggarwal C., Xin D., Shaked A., Gao M., Liu D., Borad M. J., Ramanathan R. K., Carpenter E. L., Ji A., de Beer M. C., de Beer F. C., Webb N. R., Beatty G. L., Nature 2019, 567, 249. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 2022YFA1105300/National Key Research and Development Program of China
- 81822032/National Natural Science Foundation of China
- 91959111/National Natural Science Foundation of China
- 81872027/National Natural Science Foundation of China
- 81902931/National Natural Science Foundation of China
- 82273484/National Natural Science Foundation of China
- 81874075/National Natural Science Foundation of China
- 81972305/National Natural Science Foundation of China
- 82170594/National Natural Science Foundation of China
- 82273431/National Natural Science Foundation of China
- 82003111/National Natural Science Foundation of China
- CSTC2019JCYJJQX0027/Natural Science Foundation of Chongqing Municipality
- CSTC2018JCYJAX0433/Natural Science Foundation of Chongqing Municipality
- 2019CXLCA001/Army Medical University
- 2018XLC2023/Army Medical University
- 2019XQY19/Army Medical University
- 2019XLC1012/Army Medical University
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous