

Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity
- PMID: 37829001
- PMCID: PMC10569228
- DOI: 10.1038/s42256-023-00694-6
Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity
Abstract
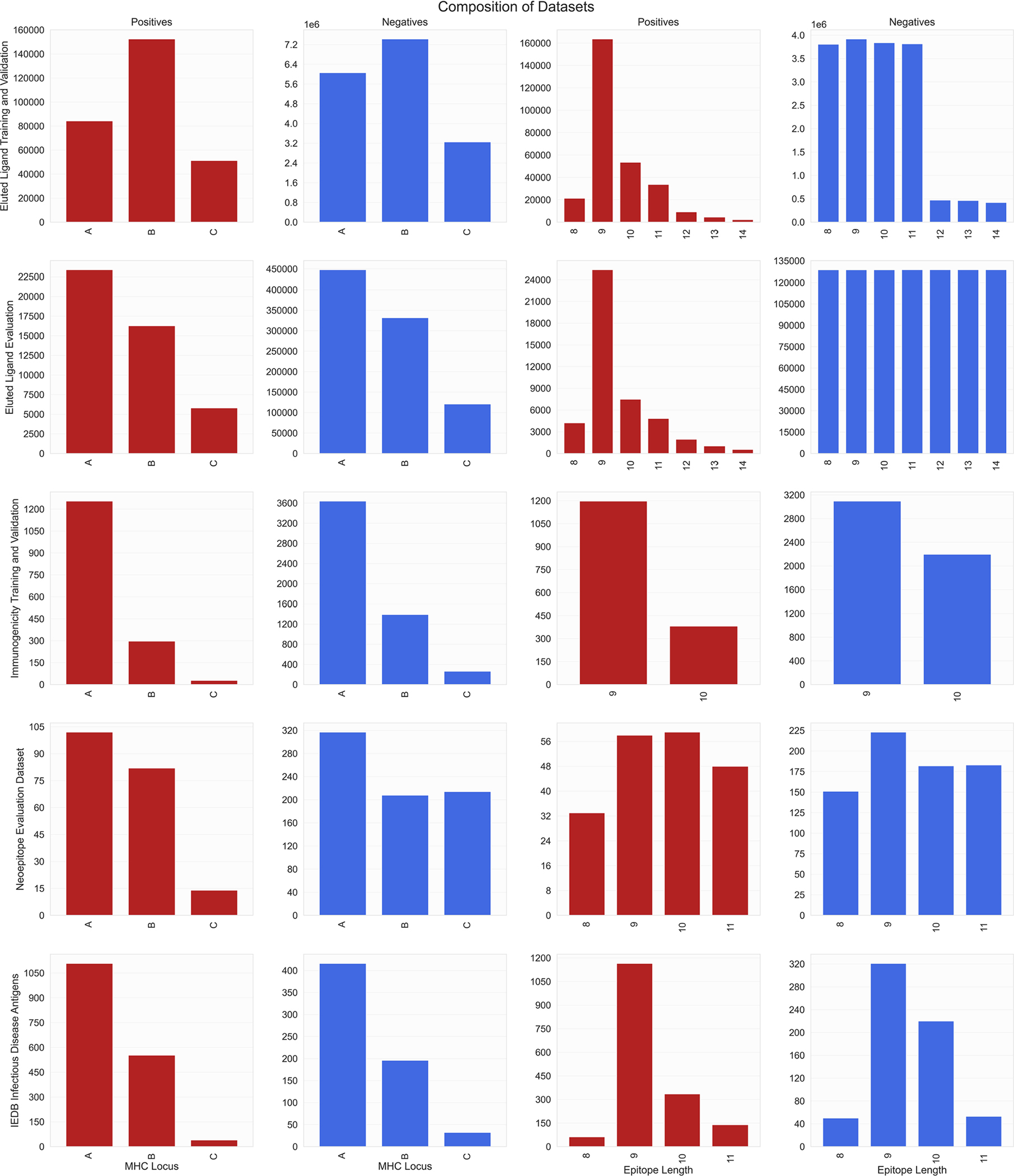
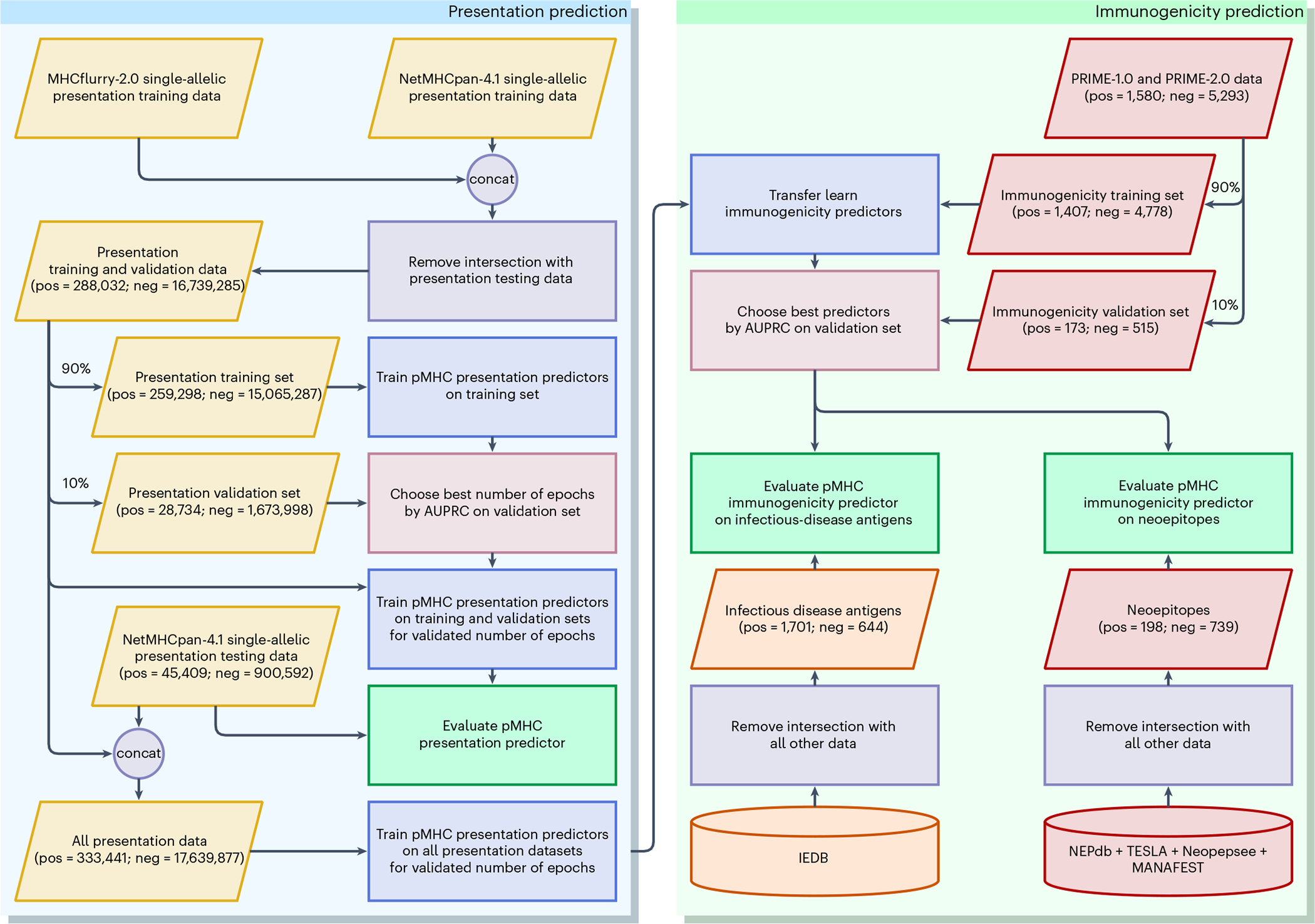
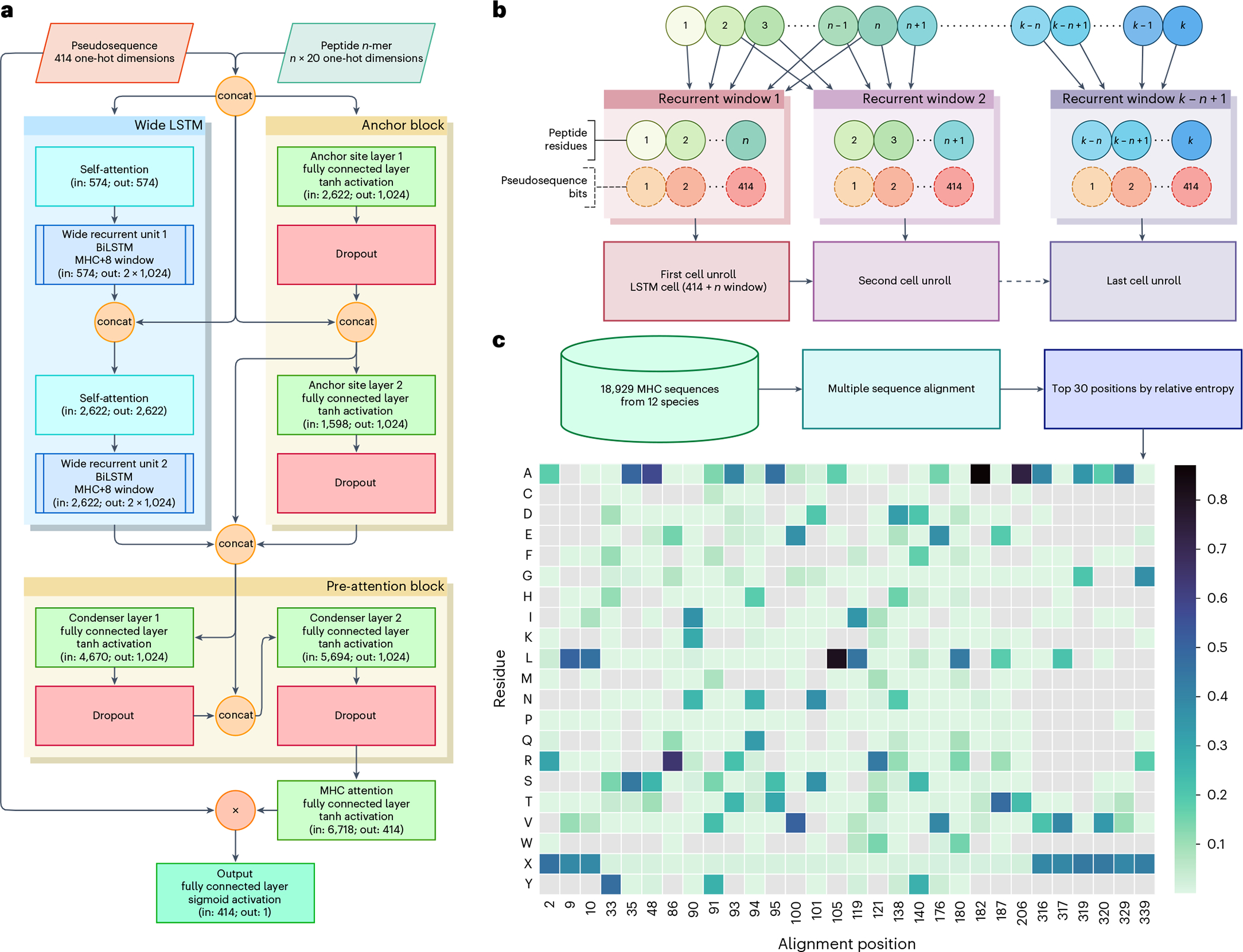
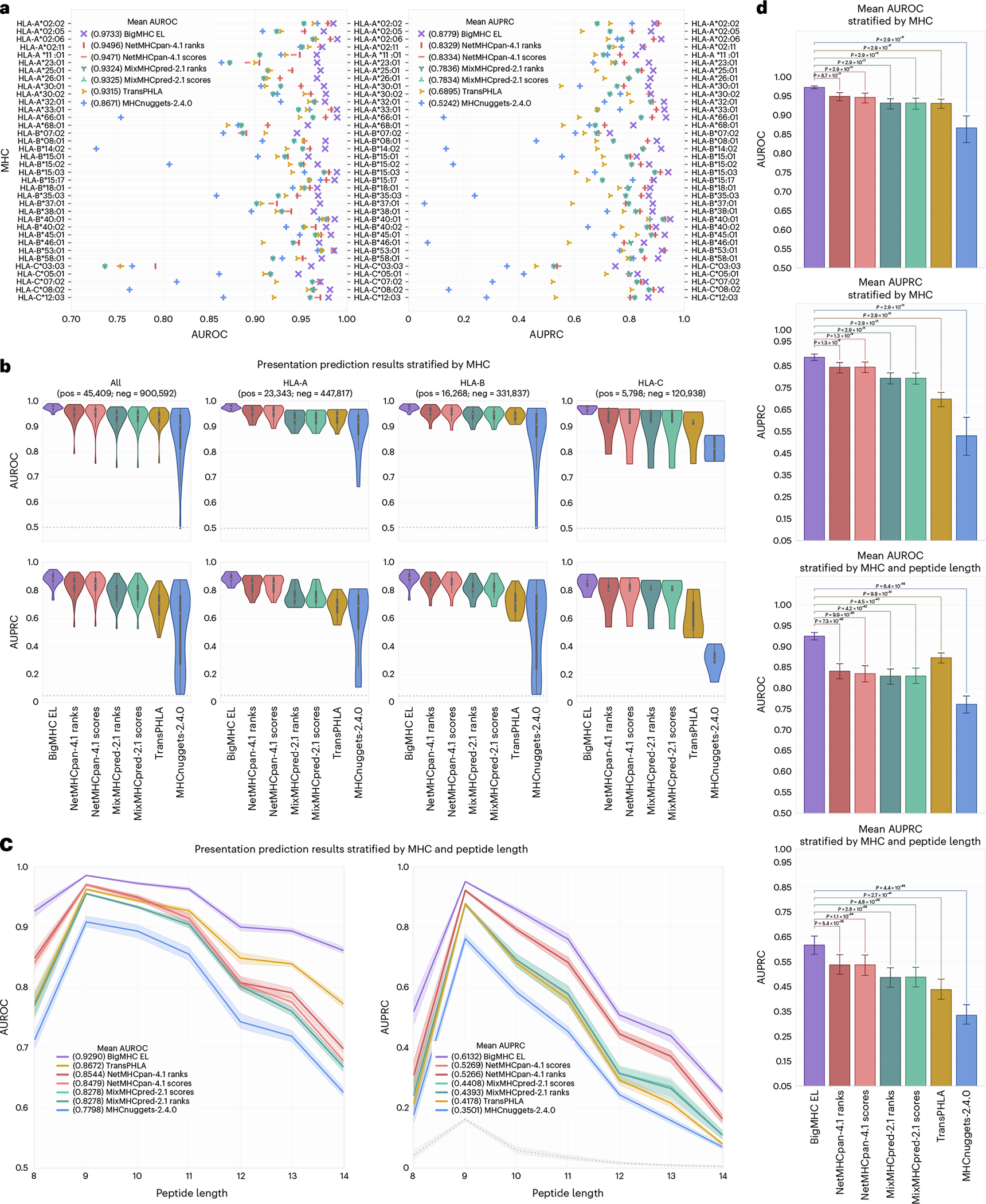
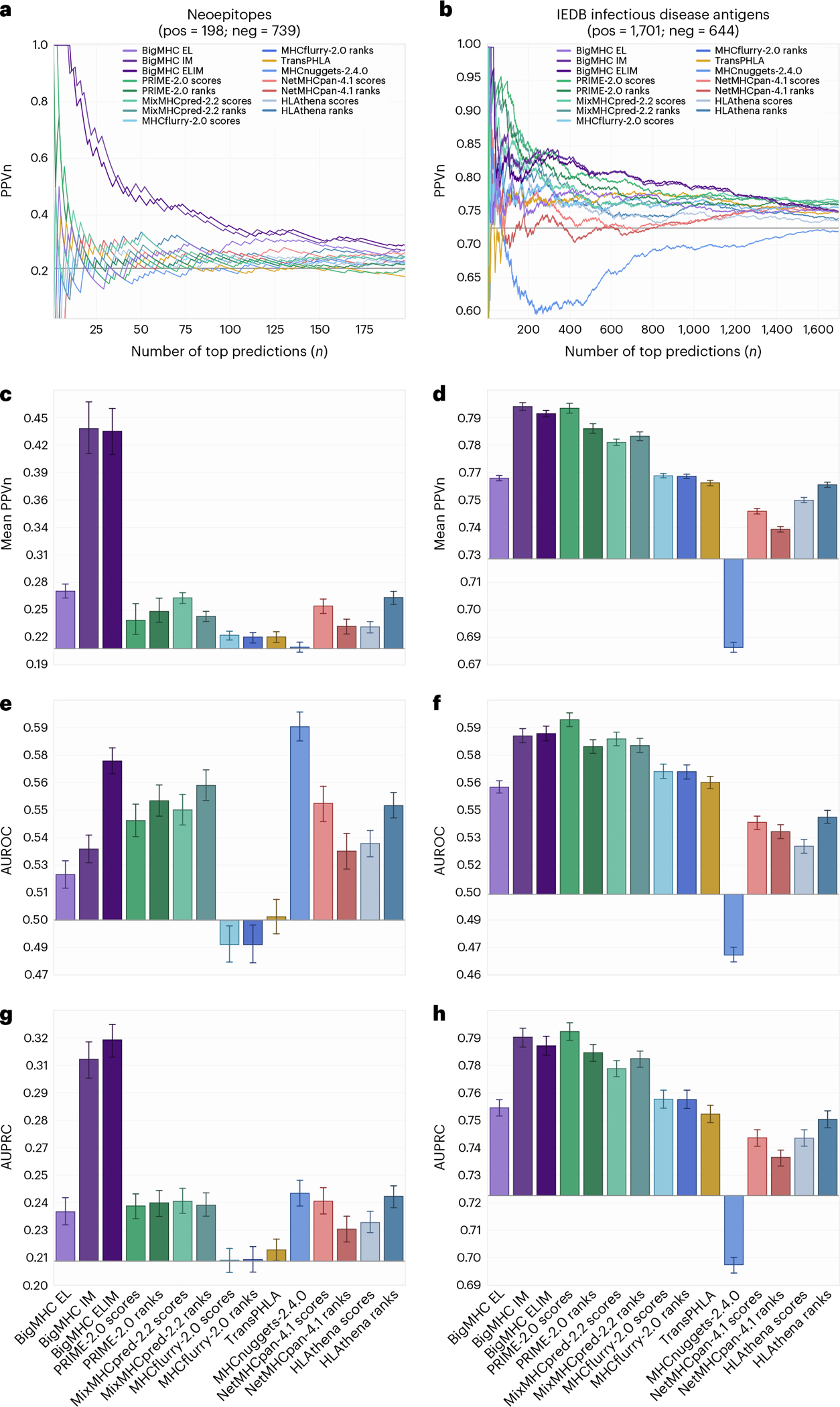
Identifying neoepitopes that elicit an adaptive immune response is a major bottleneck to developing personalized cancer vaccines. Experimental validation of candidate neoepitopes is extremely resource intensive and the vast majority of candidates are non-immunogenic, creating a needle-in-a-haystack problem. Here we address this challenge, presenting computational methods for predicting class I major histocompatibility complex (MHC-I) epitopes and identifying immunogenic neoepitopes with improved precision. The BigMHC method comprises an ensemble of seven pan-allelic deep neural networks trained on peptide-MHC eluted ligand data from mass spectrometry assays and transfer learned on data from assays of antigen-specific immune response. Compared with four state-of-the-art classifiers, BigMHC significantly improves the prediction of epitope presentation on a test set of 45,409 MHC ligands among 900,592 random negatives (area under the receiver operating characteristic = 0.9733; area under the precision-recall curve = 0.8779). After transfer learning on immunogenicity data, BigMHC yields significantly higher precision than seven state-of-the-art models in identifying immunogenic neoepitopes, making BigMHC effective in clinical settings.
Figures
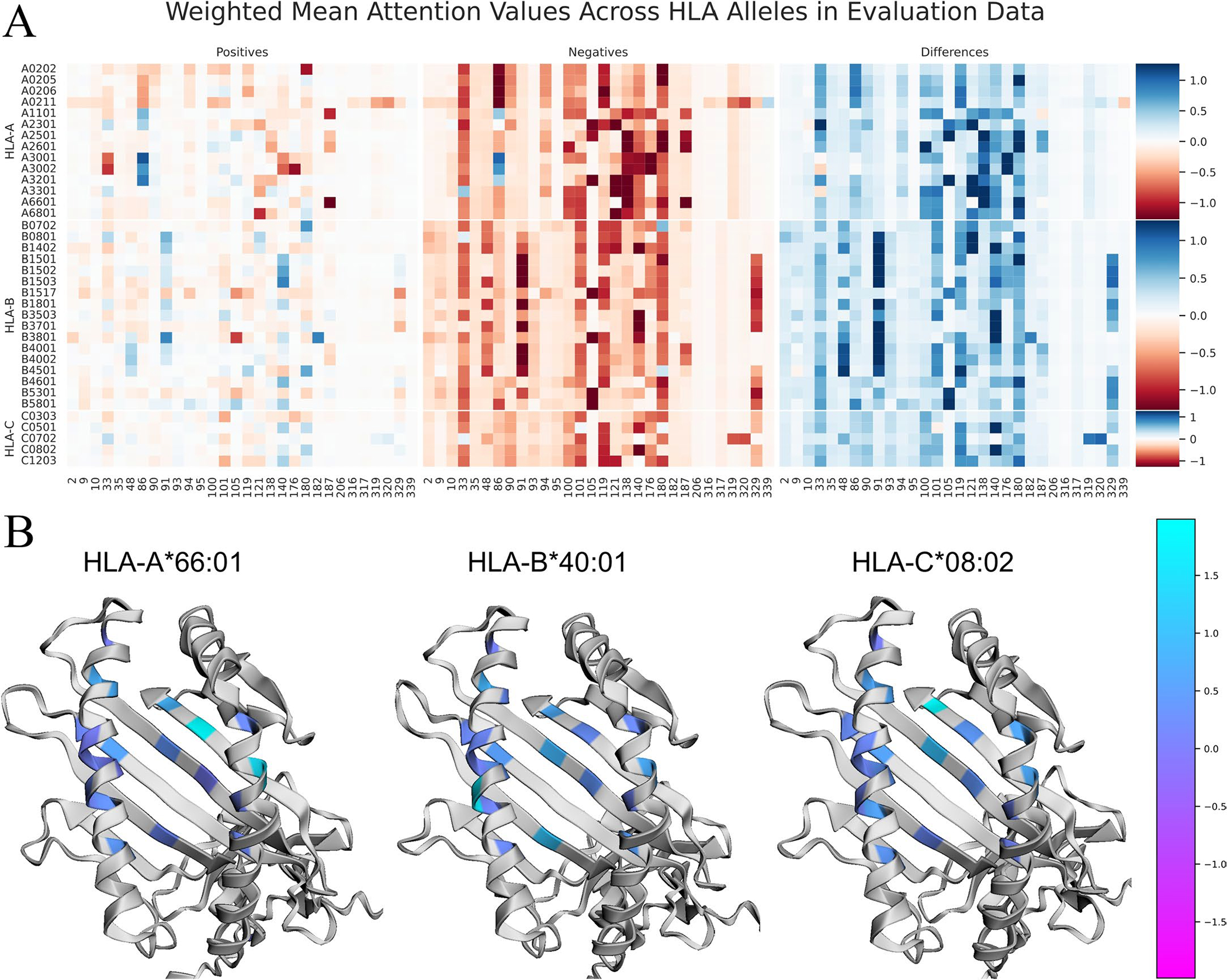
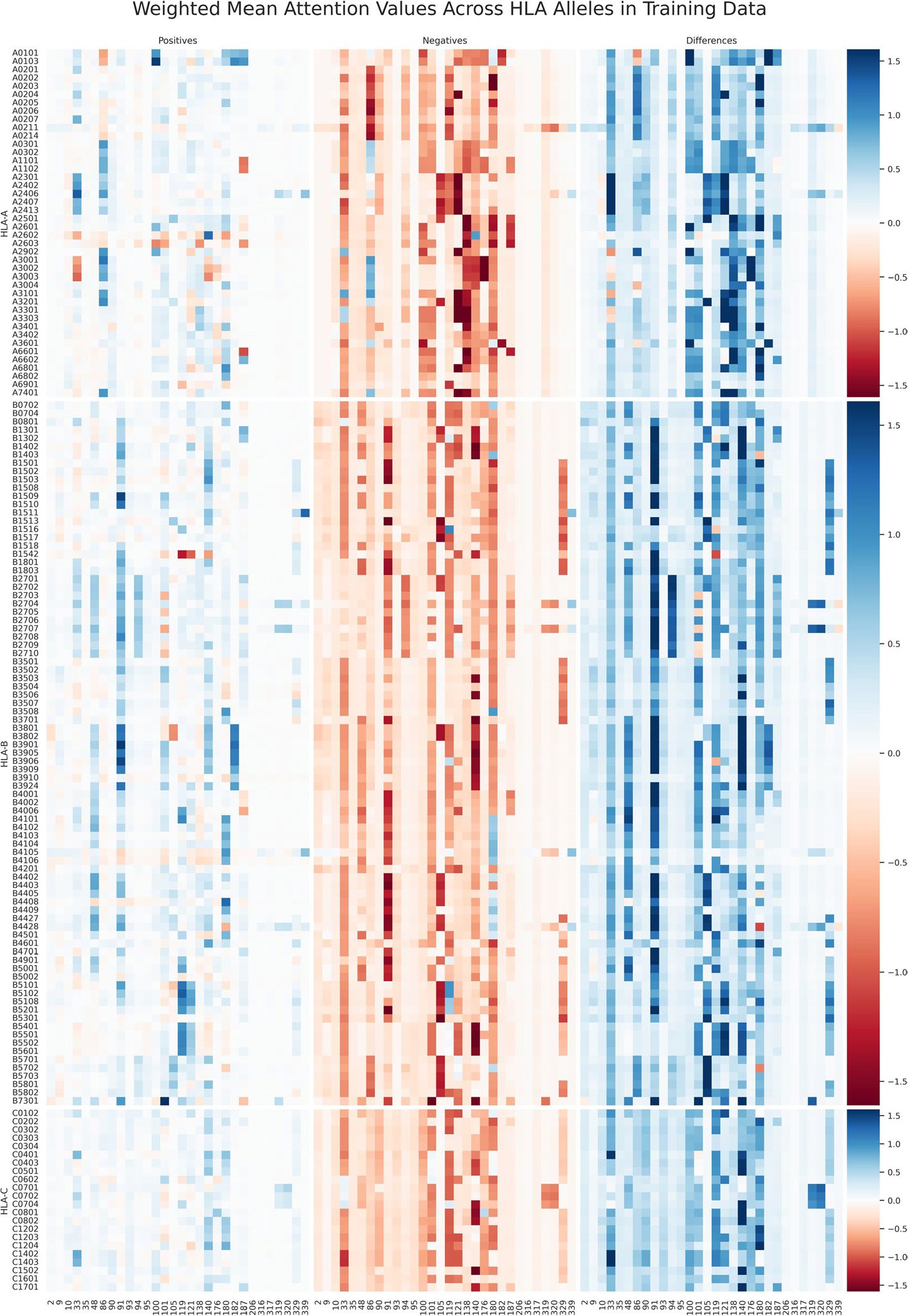
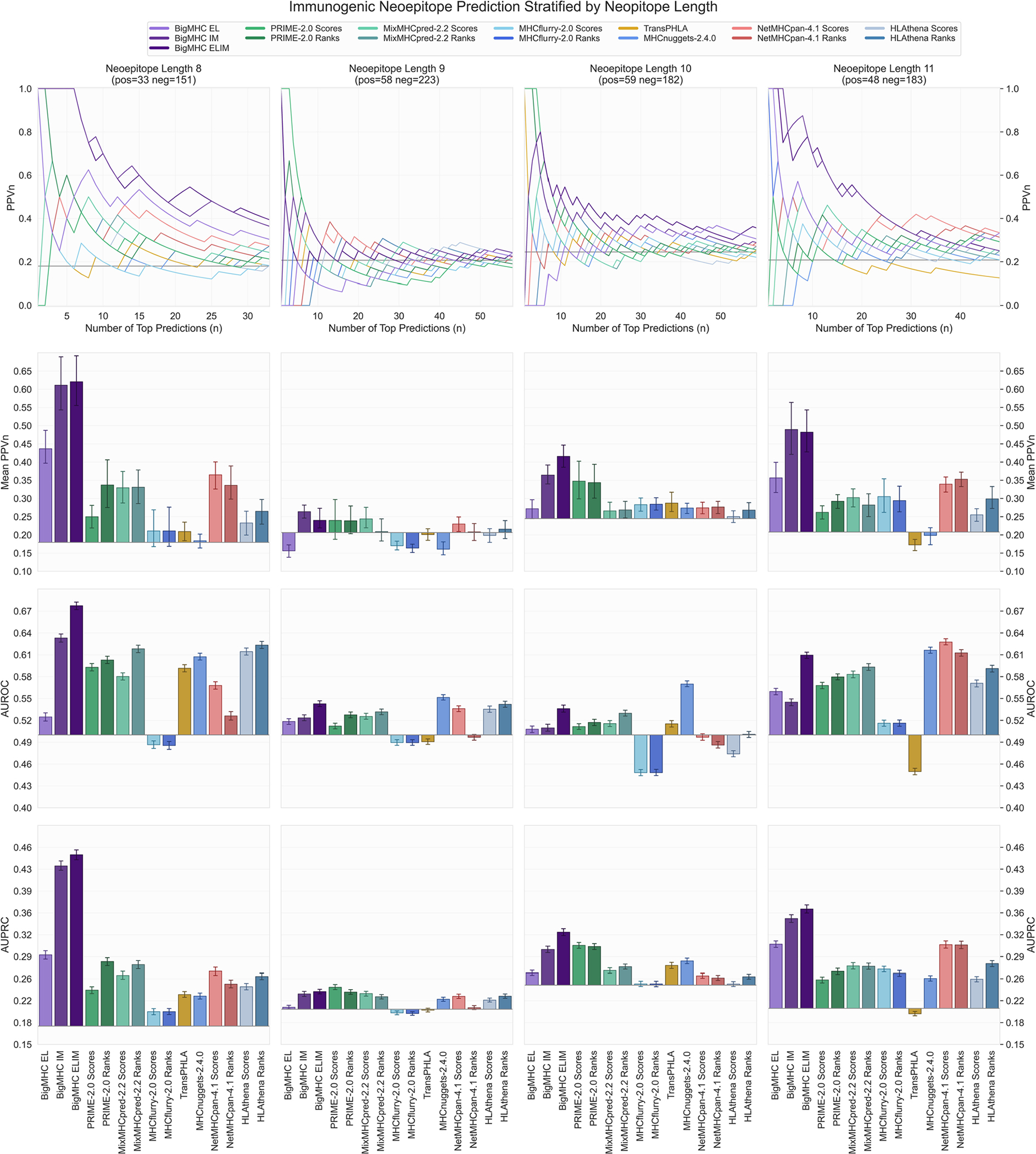
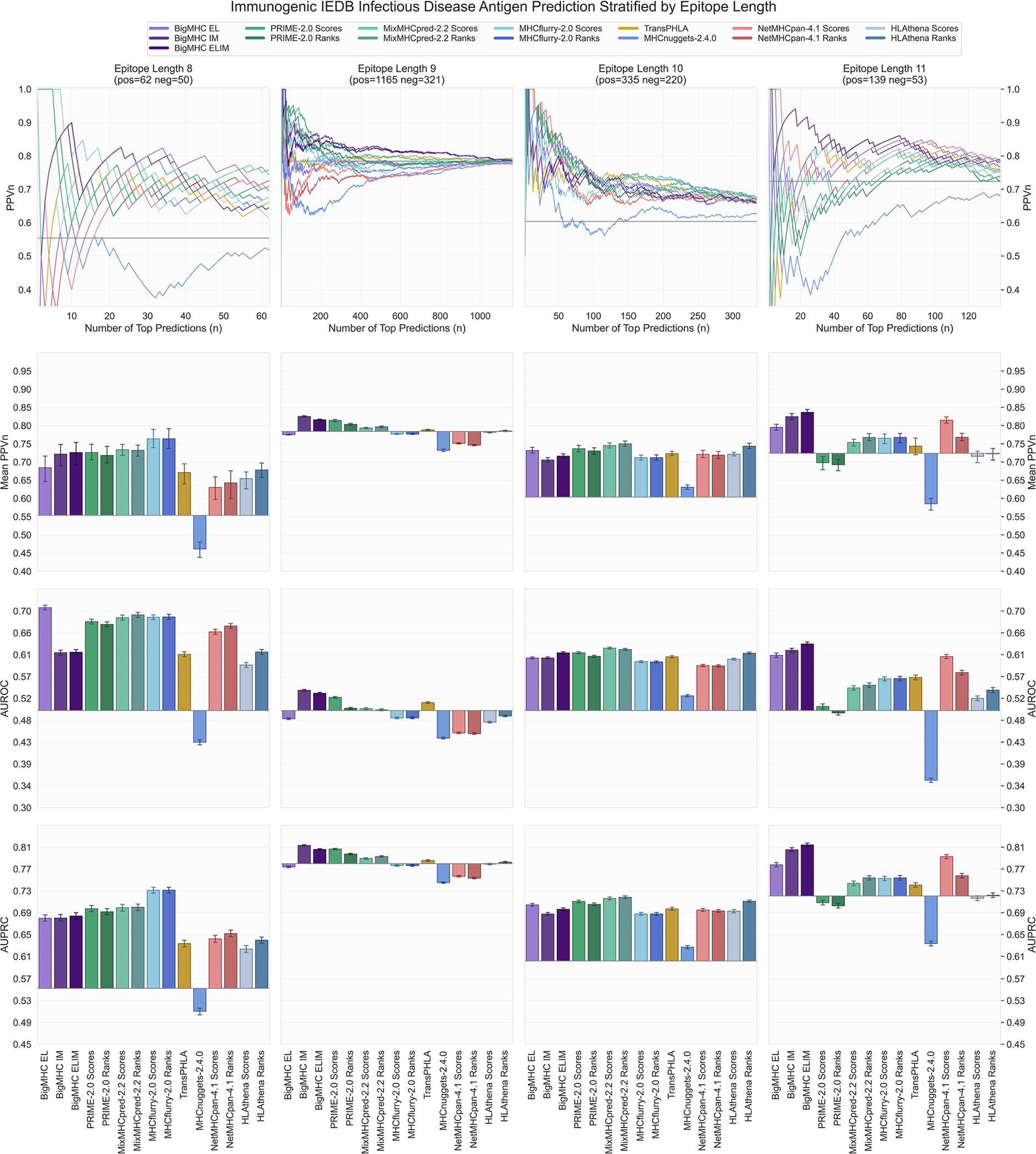
References
-
- O’Donnell TJ, Rubinsteyn A & Laserson U MHCflurry 2.0: improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 11, 42–48 (2020). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials