

Bone health in children with Angelman syndrome at the ENCORE Expertise Center
- PMID: 37831301
- PMCID: PMC10857954
- DOI: 10.1007/s00431-023-05231-6
Bone health in children with Angelman syndrome at the ENCORE Expertise Center
Abstract
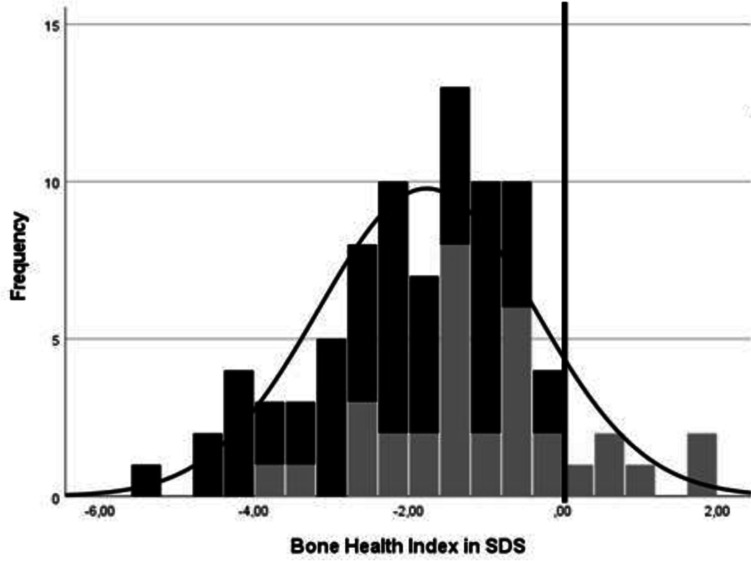
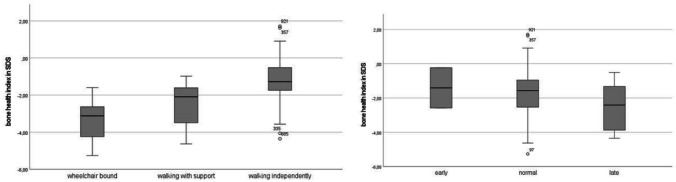
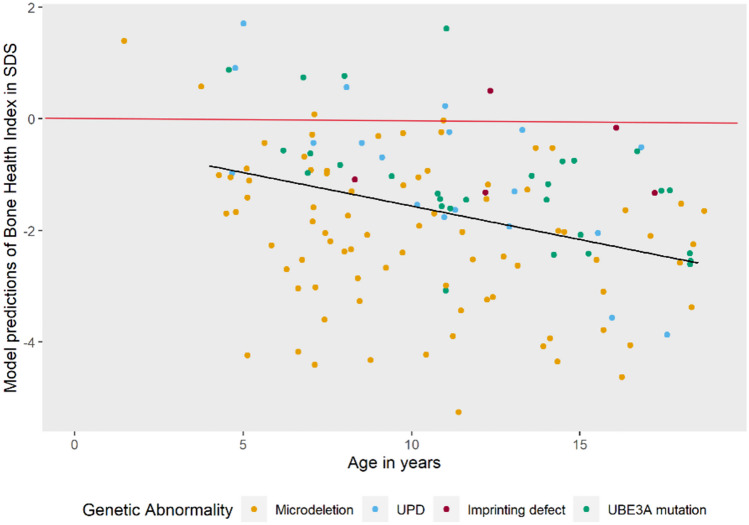
Angelman syndrome (AS) is a rare genetic disorder due to lack of UBE3A function on chromosome 15q11.2q13 caused by a deletion, uniparental paternal disomy (UPD), imprinting center disorder (ICD), or pathological variant of the UBE3A gene. AS is characterized by developmental delay, epilepsy, and lack of speech. Although fractures are observed frequently in our clinical practice, there are few studies on bone health in AS. The aim of this study is to investigate bone health in children with AS. In this prospective cohort study, we describe bone health in 91 children with AS visiting the ENCORE Expertise Center for AS between April 2010 and December 2021. Bone health was assessed with the bone health index (BHI) in standard deviation score (SDS) measured by digital radiogrammetry of the left hand using BoneXpert software. Risk factors analyzed were age, sex, genetic subtype, epilepsy, anti-seizure medication use, mobility, body mass index (BMI), and onset of puberty. Children with AS had a mean BHI of -1.77 SDS (SD 1.4). A significantly lower BHI was found in children with a deletion (-2.24 SDS) versus non-deletion (-1.02 SDS). Other factors associated with reduced BHI-SDS were inability to walk and late onset of puberty. Children with a history of one or more fractures (22%) had a significantly lower BHI than children without fractures (-2.60 vs -1.56 SDS). Longitudinal analysis showed a significant decrease in BHI-SDS with age in all genetic subtypes. Conclusions: Children with AS have a reduced bone health. Risk factors are deletion genotype, no independent walking, and late onset of puberty. Bone health decreased significantly with age. What is Known: • Children with neurological disorders often have a low bone health and higher risk of fractures. • Little is known about bone health in children with Angelman syndrome (AS). What is New: • Children with AS showed a reduced bone health and this was significantly associated with having a deletion, not being able to walk independently, and late onset of puberty. • Longitudinal analysis showed a significant decrease in bone health as children got older.
Keywords: Angelman syndrome; Bone health; Bone Health Index (BHI); Genotype; Longitudinal; Mobility.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Mertz LG, Christensen R, Vogel I, Hertz JM, Nielsen KB, Grønskov K, Østergaard JR (2013) Angelman syndrome in Denmark. Birth incidence, genetic findings, and age at diagnosis. Am J Med Genet A 161A(9):2197-203 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
