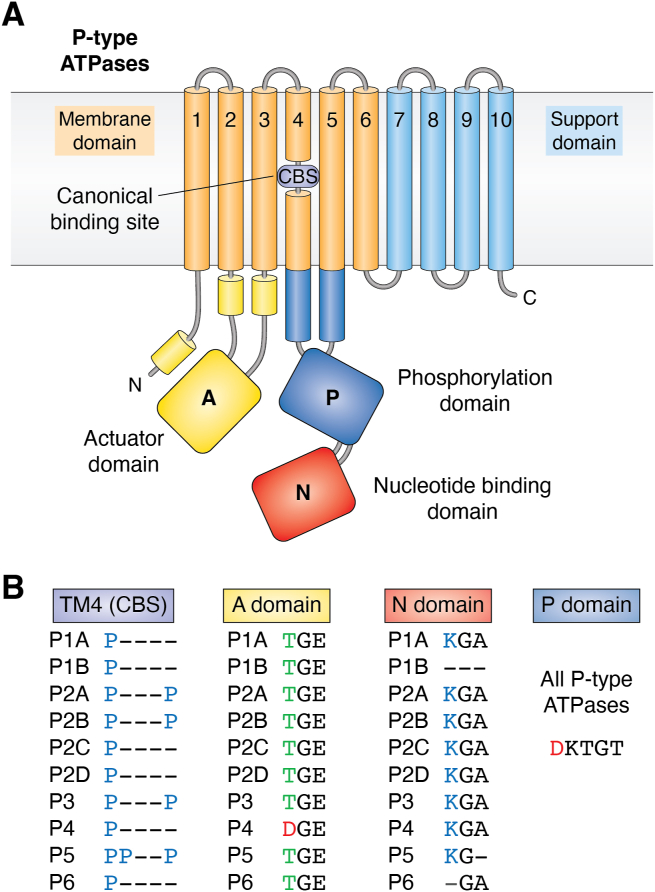
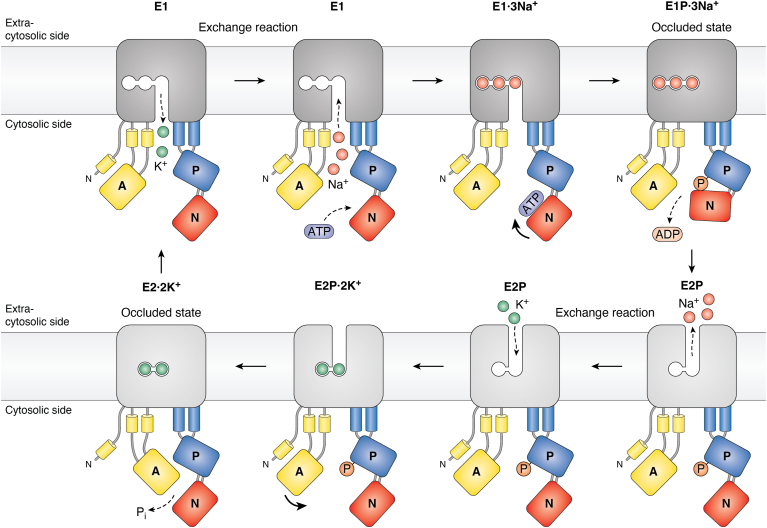
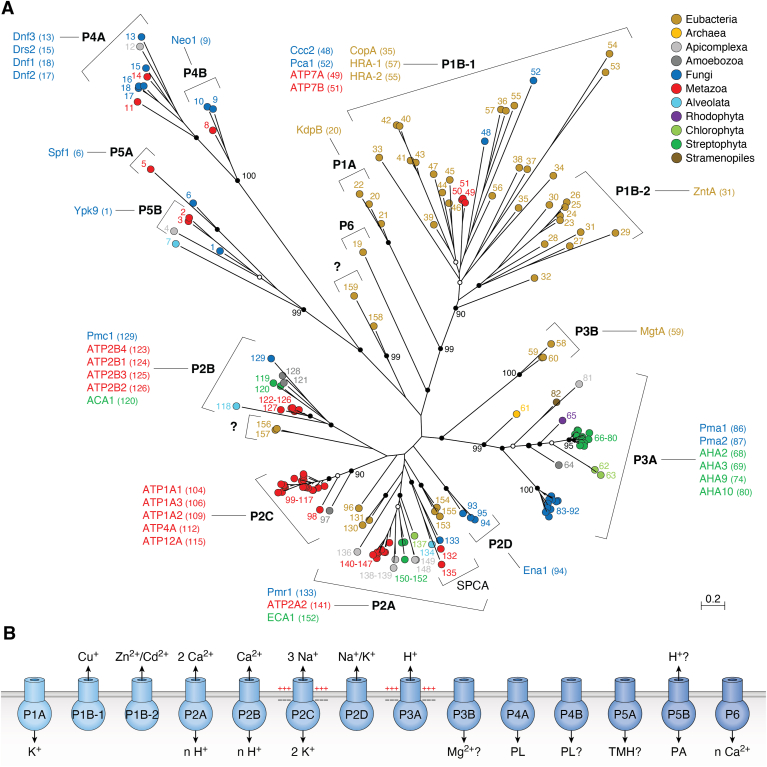
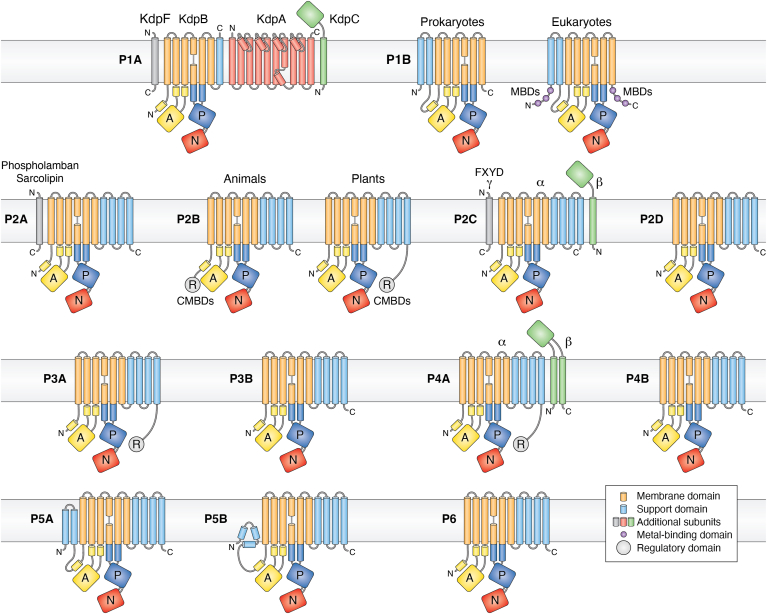
Figure 3
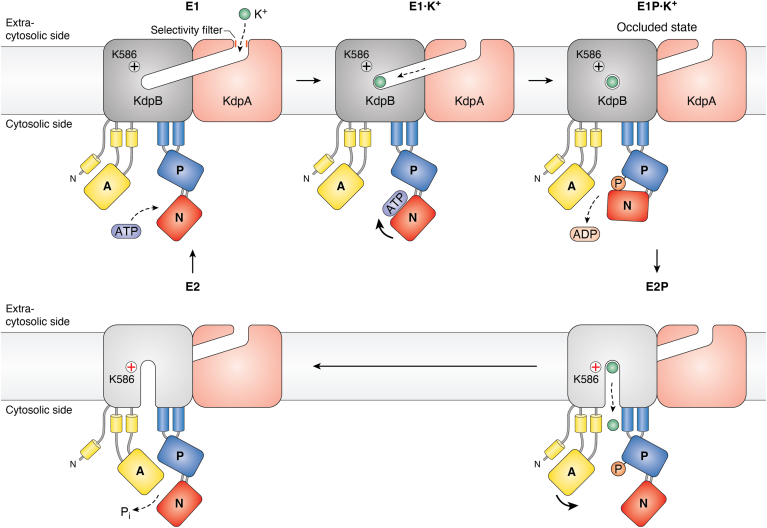
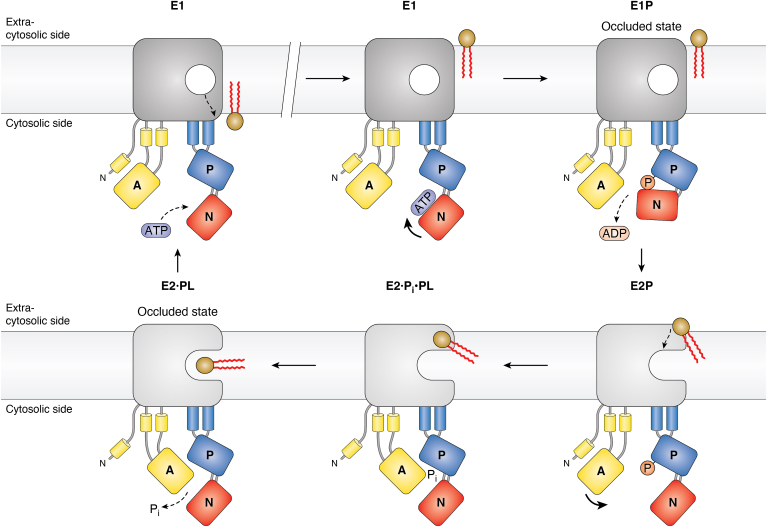
Evolution and function of P-type ATPases.A, phylogenetic tree based on core sequences of 159 P-type ATPases. The sequences used and their numbers used for constructing the phylogenetic tree are the same as in Axelsen and Palmgren (1998). Clades that represent different P-type ATPase families and sub-families are named P1A to P6. A question mark indicates that it is uncertain to which family a sequence(s) belong. The tree was constructed using the maximum likelihood method using the program RAxML and Bayesian inference analysis using the program MrBayes as described (Palmgren, 2023). Shown is the best RAxML tree after 1000 bootstrap rounds. Number at major nodes indicate bootstrap values of ≥90. A separate Bayesian inference analysis was carried out, which resulted in a similar tree. The Bayesian inference analysis was run for 3,000,000 generations with a resulting average standard deviation of split frequencies at 0.01. Black dots at nodes in the RAxML tree indicate maximum statistical support (p = 1) in the Bayesian inference analysis; open circles at nodes indicate that p ≥ 0.95. Names of some proteins from Homo sapiens, the yeast Saccharomyces cerevisiae and the plant Arabidopsis thaliana are given before numbers. Numbers refer to sequences as follows: 1, Q12697 Ypk9 (Saccharomyces cerevisiae); 2, Q27533 W08D2.5 (Caenorhabditis elegans); 3, Q21286 catp-5 (C. elegans); 4, Q04956 Probable cation-transporting ATPase 1 (Plasmodium falciparum); 5, P90747 catp-8 (C. elegans); 6, P39986 Spf1 (S. cerevisiae); 7, Q95050 TPA9 (Tetrahymena thermophila); 8, G5EBH1 tat-5 (C. elegans); 9, P40527 Neo1 (S. cerevisiae); 10, Q10309 neo1 (Schizosaccharomyces pombe); 11, Q7JPE3 tat-4 (C. elegans); 12, Q27720 Phospholipid-transporting ATPase (P. falciparum); 13, Q12674 Dnf3 (S. cerevisiae); 14, Q29449 ATP8A1 (Bos taurus); 15, P39524 Drs2 (S. cerevisiae); 16, Q09891 dnf2 (S. pombe); 17, Q12675 Dnf2 (S. cerevisiae); 18, P32660 Dnf1 (S. cerevisiae); 19, P9WPT1 ctpE (Mycobacterium tuberculosis); 20, P03960 KdpB (Escherichia coli); 21, P9WPU3 KdpB (M. tuberculosis); 22, P73867 KdpB (Synechocystis sp. PCC 6803 substr. Kazusa); 23, P94888 cadA (Lactococcus lactis); 24, Q60048 cadA (Listeria monocytogenes); 25, P30336 cadA (Alkalihalophilus pseudofirmus); 26, P37386 cadA (Staphylococcus aureus); 27, Q59998 ziaA (Synechocystis sp.); 28, Q59997 slr0797 (Synechocystis sp.); 29, Q59465 cadA (Helicobacter pylori); 30, P9WPS7 ctpG (M. tuberculosis); 31, P37617 zntA (E. coli); 32, P9WPT5 ctpC (M. tuberculosis); 33, P77871 copA (H. pylori); 34, P77868 HI_0290 (Haemophilus influenzae); 35, Q59385 copA (E. coli); 36, P05425 copB (Enterococcus hirae); 37, P37385 synA (Synechococcus elongatus); 38, P74512 Cation-transporting ATPase (Synechocystis sp.); 39, P9WPS3 ctpV (M. tuberculosis); 40, P46840 ctpB (Mycobacterium leprae); 41, P9WPU1 ctpA (M. tuberculosis); 42, P9WPT9 ctpB (M. tuberculosis); 43, P46839 ctpA (M. leprae); 44, Q59688 copA_3 (Proteus mirabilis); 45, P73241 pacS (Synechocystis sp.); 46, P37279 pacS (S. elongatus); 47, P77881 ctpA (L. monocytogenes); 48, P38995 Ccc2 (S. cerevisiae); 49, Q04656 ATP7A (Homo sapiens); 50, Q64535 Atp7b (R. norvegicus); 51, P35670 ATP7B (H. sapiens); 52, P38360 Pca1 (S. cerevisiae); 53, Q59207 fixI (Bradyrhizobium diazoefficiens); 54, P18398 fixI (Rhizobium meliloti); 55, Q59370 HRA-2 (E. coli); 56, P32113 copA (E. hirae); 57, Q59369 HRA-1 (E. coli); 58, P22036 mgtB (Salmonella typhimurium); 59, P0ABB8 mgtA (E. coli); 60, P36640 mgtA (S. typhimurium); 61, Q58623 MJ1226 (Methanocaldococcus jannaschii); 62, P54210 DHA1 (Dunaliella acidophila); 63, P54211 PMA1 (D. bioculata); 64, P54679 patB (Dictyostelium discoideum); 65, O04956 Plasma membrane ATPase (Cyanidium caldarium); 66, Q43178 PHA2 (Solanum tuberosum); 67, Q03194 PMA4 (Nicotiana plumbaginifolia); 68, P19456 AHA2 (A. thaliana); 69, P20431 AHA3 (A. thaliana); 70, Q43131 Plasma membrane ATPase (Vicia faba); 71, Q43275 zha1 (Zostera marina); 72, Q43271 MHA2 (Zea mays); 73, P93265 PMA (Mesembryanthemum crystallinum); 74, Q42556 AHA9 (A. thaliana); 75, Q43002 OSA2 (Oryza sativa); 76, Q43243 MHA1 (Z. mays); 77, Q43001 OSA1 (O. sativa); 78, Q42932 Plasma membrane ATPase (N. plumbaginifolia); 79, Q43106 BHA-1 (Phaseolus vulgaris); 80, Q43128 AHA10 (A. thaliana); 81, P12522 H1B (Leishmania donovani); 82, A0A7S3XUR3 Plasma membrane ATPase (Heterosigma akashiwo); 83, P24545 PMA1 (Zygosaccharomyces rouxii); 84, P28877 PMA1 (Candida albicans); 85, P49380 PMA1 (Kluyveromyces lactis); 86, P05030 Pma1 (S. cerevisiae); 87, P19657 Pma2 (S. cerevisiae); 88, Q92446 PCA1 (Pneumocystis carinii); 89, P28876 PMA2 (S. pombe); 90, P09627 PMA1 (S. pombe); 91, P07038 pma-1 (Neurospora crassa); 92, Q07421 PMA1 (Ajellomyces capsulatus); 93, P22189 cta3 (S. pombe); 94, P13587 Ena1 (S. cerevisiae); 95, P78981 Z-ENA1 (Zygosaccharomyces rouxii); 96, P73273 ziaA (Synechocystis sp.); 97, Q76P11 ionA (D. discoideum); 98, G5EFV6 catp-4 (C. elegans); 99, P35317 ATP1A (Hydra vulgaris); 100, P28774 Na+/K+-ATPase alpha-B (Artemia franciscana); 101, Q27461 eat-6 (C. elegans); 102, P13607 Atpalpha (Drosophila melanogaster); 103, Q27766 Na+/K+-ATPase alpha (Ctenocephalides felis); 104, P05023 ATP1A1 (H. sapiens); 105, P05025 ATP1A (Tetronarce californica); 106, P13637 ATP1A3 (H. sapiens); 107, Q92030 atp1a1 (Anguilla anguilla); 108, P25489 atp1a1 (Catostomus commersonii); 109, P50993 ATP1A2 (H. sapiens); 110, Q64541 Atp1a4 (R. norvegicus); 111, P17326 Na+/K+-ATPase alpha-A (A. franciscana); 112, P20648 ATP4A (H. sapiens); 113, Q92126 atp4a (Xenopus laevis); 114, Q64392 ATP12A (Cavia porcellus); 115, P54707 ATP12A (H. sapiens); 116, P54708 Atp12A (R. norvegicus); 117, Q92036 ATP12A (Rhinella marina); 118, Q27829 Plasma membrane calcium ATPase (Paramecium tetraurelia); 119, P93067 Calcium-transporting ATPase (Brassica oleracea); 120, Q37145 ACA1 (A. thaliana); 121, Q27642 Calcium-transporting ATPase (Entamoeba histolytica); 122, Q64542 Atp2b4 (R. norvegicus); 123, P23634 ATP2B4 (H. sapiens); 124, P20020 ATP2B1 (H. sapiens); 125, Q16720 ATP2B3 (H. sapiens); 126, Q01814 ATP2B2 (H. sapiens); 127, G5EFR6 mca-1 (C. elegans); 128, P54678 patA (D. discoideum);129, P38929 Pmc1 (S. cerevisiae); 130, P9WPS9 ctpF (M. tuberculosis); 131, P37367 pma1 (Synechocystis sp.); 132, Q64566 Atp2c1 (R. norvegicus); 133, P13586 Pmr1 (S. cerevisiae); 134, Q95022 CppA-E1 (Cryptosporidium parvum); 135, Q27724 PfATPase4 (P. falciparum); 136, Q95060 TVCA1 (Trichomonas vaginalis); 137, P54209 CA1 (Dunaliella bioculata); 138, O09489 Calcium-transporting ATPase (Leishmania amazonensis); 139, P35315 TBA1 (Trypanosoma brucei brucei); 140, Q27779 SMA1 (Schistosoma mansoni); 141, P16615 ATP2A2 (H. sapiens); 142, P70083 atp2a1 (Makaira nigricans); 143, Q92105 ATP2A1 (Pelophylax lessonae); 144, Q64578 Atp2a1 (Rattus norvegicus); 145, P18596 Atp2a3 (R. norvegicus); 146, P22700 SERCA (D. melanogaster); 147, P35316 SERCA (A. franciscana); 148, Q08853 ATP6 (P. falciparum); 149, Q27764 YEL6 (Plasmodium yoelii); 150, Q42883 LCA1 (Solanum lycopersicum); 151, O04938 Ca2+-ATPase (O. sativa); 152, P92939 ECA1 (A. thaliana); 153, Q59999 sll0672 (Synechocystis sp.); 154, P37278 pacL (S. elongatus); 155, P74062 slr0822 (Synechocystis sp.); 156, P78036 pacL (Mycoplasma pneumoniae); 157, P47317 pacL (Mycoplasma genitalium); 158, P9WPS5 ctpI (M. tuberculosis); 159, P96271 ctpH (M. tuberculosis). Scale bar, 0.2 amino acid substitutions per site. B, overview of P-type ATPase families and transported ligands. Domain and subunit organization are not shown. For each subfamily is shown the ligand transported, the number of ligands transported per ATP hydrolyzed, and the direction of transport. Not shown are subunit and transmembrane helices. A subgroup of P2A ATPases, secretory pathway Ca2+-ATPases (marked SPCA in Fig. 3A), only transport one Ca2+ per ATP hydrolyzed. Depending on the stoichiometry of transport, several P-type ATPases can be electrogenic. P2C and P3A ATPases are highly electrogenic and maintain plasma membrane potentials that are negative on the cytosolic side of the membrane. Abbreviations: PA, polyamines; PL, phospholipids; TMH, transmembrane helices.