

A homozygous missense variant in the YG box domain in an individual with severe spinal muscular atrophy: a case report and variant characterization
- PMID: 37841286
- PMCID: PMC10571918
- DOI: 10.3389/fncel.2023.1259380
A homozygous missense variant in the YG box domain in an individual with severe spinal muscular atrophy: a case report and variant characterization
Abstract
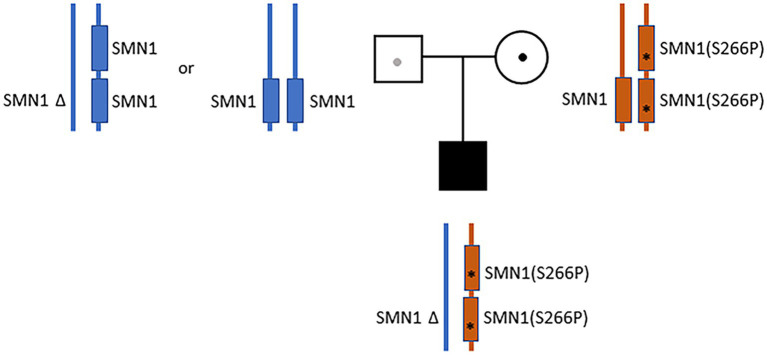
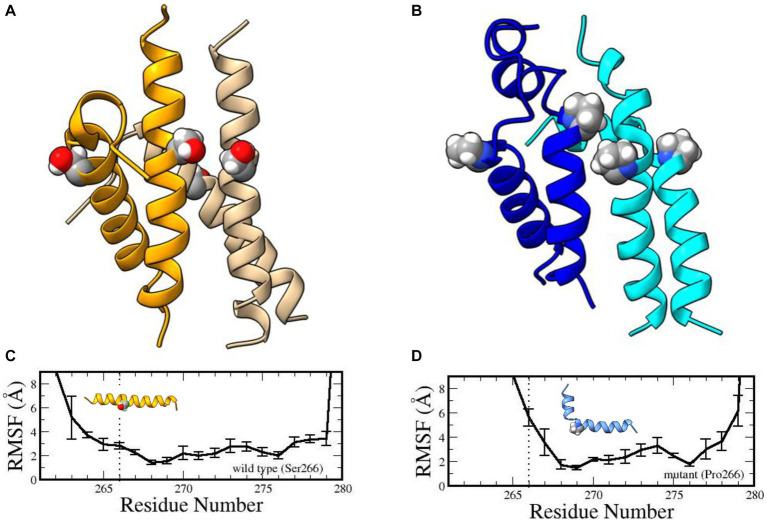
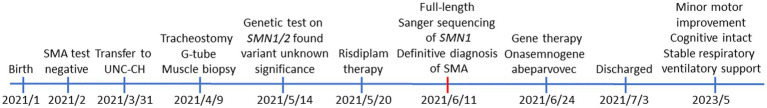
The vast majority of severe (Type 0) spinal muscular atrophy (SMA) cases are caused by homozygous deletions of survival motor neuron 1 (SMN1). We report a case in which the patient has two copies of SMN1 but clinically presents as Type 0 SMA. The patient is an African American male carrying a homozygous maternally inherited missense variant (c.796T>C) in a cis-oriented SMN1 duplication on one chromosome and an SMN1 deletion on the other chromosome (genotype: 2*+0). Initial extensive genetic workups including exome sequencing were negative. Deletion analysis used in the initial testing for SMA also failed to detect SMA as the patient has two copies of SMN1. Because of high clinical suspicion, SMA diagnosis was finally confirmed based on full-length SMN1 sequencing. The patient was initially treated with risdiplam and later gene therapy with onasemnogene abeparvovec at 5 months without complications. The patient's muscular weakness has stabilized with mild improvement. The patient is now 28 months old and remains stable and diffusely weak, with stable respiratory ventilatory support. This case highlights challenges in the diagnosis of SMA with a non-deletion genotype and provides a clinical example demonstrating that disruption of functional SMN protein polymerization through an amino acid change in the YG-box domain represents a little known but important pathogenic mechanism for SMA. Clinicians need to be mindful about the limitations of the current diagnostic approach for SMA in detecting non-deletion genotypes.
Keywords: African American; YG Box; c.796T>C variant; g.27134T>G polymorphism; modeling; non-deletion; spinal muscular atrophy (SMA).
Copyright © 2023 Li, Perera, Varghese, Shiloh-Malawsky, Hunter, Sneddon, Powell, Matera and Fan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
References
-
- Alias L., Bernal S., Fuentes-Prior P., Barcelo M. J., Also E., Martinez-Hernandez R., et al. . (2009). Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum. Genet. 125, 29–39. doi: 10.1007/s00439-008-0598-1, PMID: - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources