

Mitochondrial dysfunction and quality control lie at the heart of subarachnoid hemorrhage
- PMID: 37843218
- PMCID: PMC10664111
- DOI: 10.4103/1673-5374.381493
Mitochondrial dysfunction and quality control lie at the heart of subarachnoid hemorrhage
Abstract
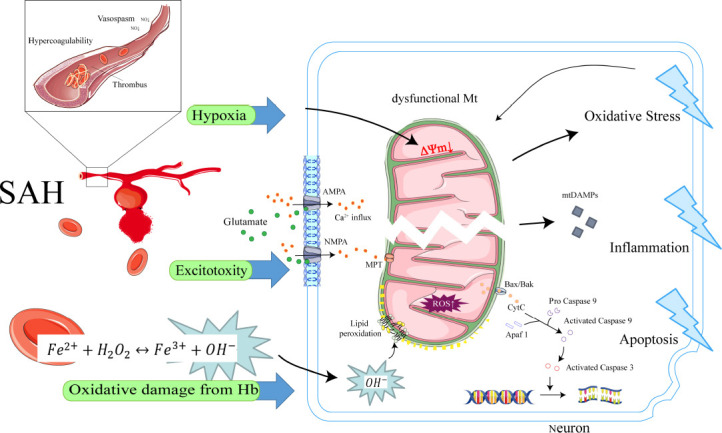
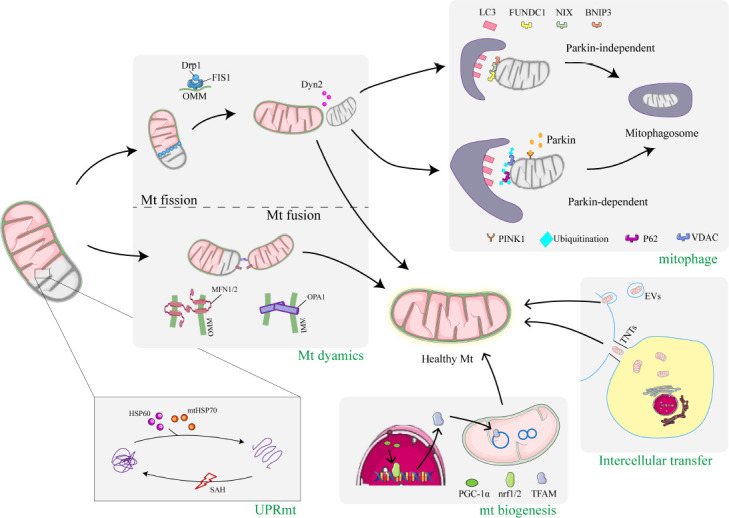
The dramatic increase in intracranial pressure after subarachnoid hemorrhage leads to a decrease in cerebral perfusion pressure and a reduction in cerebral blood flow. Mitochondria are directly affected by direct factors such as ischemia, hypoxia, excitotoxicity, and toxicity of free hemoglobin and its degradation products, which trigger mitochondrial dysfunction. Dysfunctional mitochondria release large amounts of reactive oxygen species, inflammatory mediators, and apoptotic proteins that activate apoptotic pathways, further damaging cells. In response to this array of damage, cells have adopted multiple mitochondrial quality control mechanisms through evolution, including mitochondrial protein quality control, mitochondrial dynamics, mitophagy, mitochondrial biogenesis, and intercellular mitochondrial transfer, to maintain mitochondrial homeostasis under pathological conditions. Specific interventions targeting mitochondrial quality control mechanisms have emerged as promising therapeutic strategies for subarachnoid hemorrhage. This review provides an overview of recent research advances in mitochondrial pathophysiological processes after subarachnoid hemorrhage, particularly mitochondrial quality control mechanisms. It also presents potential therapeutic strategies to target mitochondrial quality control in subarachnoid hemorrhage.
Keywords: mitochondrial biogenesis; mitochondrial dynamics; mitochondrial dysfunction; mitochondrial fission and fusion; mitochondrial quality control; mitophagy; subarachnoid hemorrhage.
Conflict of interest statement
Figures
Similar articles
-
Mitochondrial disorder and treatment of ischemic cardiomyopathy: Potential and advantages of Chinese herbal medicine.Biomed Pharmacother. 2023 Mar;159:114171. doi: 10.1016/j.biopha.2022.114171. Epub 2023 Jan 13. Biomed Pharmacother. 2023. PMID: 36641924 Review.
-
Mitochondrial biogenesis: pharmacological approaches.Curr Pharm Des. 2014;20(35):5507-9. doi: 10.2174/138161282035140911142118. Curr Pharm Des. 2014. PMID: 24606795
-
Mitochondrial Quality Control in Cerebral Ischemia-Reperfusion Injury.Mol Neurobiol. 2021 Oct;58(10):5253-5271. doi: 10.1007/s12035-021-02494-8. Epub 2021 Jul 18. Mol Neurobiol. 2021. PMID: 34275087 Review.
-
Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure.J Mol Med (Berl). 2019 Apr;97(4):451-462. doi: 10.1007/s00109-019-01756-2. Epub 2019 Feb 21. J Mol Med (Berl). 2019. PMID: 30788535 Review.
-
New Mechanisms and Targets of Subarachnoid Hemorrhage: A Focus on Mitochondria.Curr Neuropharmacol. 2022;20(7):1278-1296. doi: 10.2174/1570159X19666211101103646. Curr Neuropharmacol. 2022. PMID: 34720082 Free PMC article. Review.
Cited by
-
Bromodomain-containing protein 4 knockdown promotes neuronal ferroptosis in a mouse model of subarachnoid hemorrhage.Neural Regen Res. 2026 Feb 1;21(2):715-729. doi: 10.4103/NRR.NRR-D-24-00147. Epub 2024 Jul 29. Neural Regen Res. 2026. PMID: 39104173 Free PMC article.
-
Role of mitophagy in spinal cord ischemia-reperfusion injury.Neural Regen Res. 2026 Feb 1;21(2):598-611. doi: 10.4103/NRR.NRR-D-24-00668. Epub 2024 Dec 7. Neural Regen Res. 2026. PMID: 39665804 Free PMC article.
-
Peripheral mitochondrial DNA as a neuroinflammatory biomarker for major depressive disorder.Neural Regen Res. 2025 Jun 1;20(6):1541-1554. doi: 10.4103/NRR.NRR-D-23-01878. Epub 2024 Jun 26. Neural Regen Res. 2025. PMID: 38934398 Free PMC article.
-
NLRP3 inflammasome and gut microbiota-brain axis: A new perspective on white matter injury after intracerebral hemorrhage.Neural Regen Res. 2026 Jan 1;21(1):62-80. doi: 10.4103/NRR.NRR-D-24-00917. Epub 2025 Jan 29. Neural Regen Res. 2026. PMID: 39885662 Free PMC article.
-
The Role of Endothelial Cell Mitophagy in Age-Related Cardiovascular Diseases.Aging Dis. 2024 Jul 26;16(4):2151-2176. doi: 10.14336/AD.2024.0788. Aging Dis. 2024. PMID: 39122456 Free PMC article. Review.
References
-
- Akeret K, Buzzi RM, Schaer CA, Thomson BR, Vallelian F, Wang S, Willms J, Sebök M, Held U, Deuel JW, Humar R, Regli L, Keller E, Hugelshofer M, Schaer DJ. Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury. J Cereb Blood Flow Metab. 2021;41:3000–3015. - PMC - PubMed
-
- Andersen CR, Presseau J, Saigle V, Etminan N, Vergouwen MDI, English SW. Core outcomes for subarachnoid haemorrhage. Lancet Neurol. 2019;18:1075–1076. - PubMed
-
- Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100. - PubMed
-
- Bonora M, Giorgi C, Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol. 2022;23:266–285. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
