

Landscape of Clinical Resistance Mechanisms to FGFR Inhibitors in FGFR2-Altered Cholangiocarcinoma
- PMID: 37843855
- PMCID: PMC10767308
- DOI: 10.1158/1078-0432.CCR-23-1317
Landscape of Clinical Resistance Mechanisms to FGFR Inhibitors in FGFR2-Altered Cholangiocarcinoma
Abstract
Purpose: FGFR inhibitors are effective in FGFR2-altered cholangiocarcinoma, leading to approval of reversible FGFR inhibitors, pemigatinib and infigratinib, and an irreversible inhibitor, futibatinib. However, acquired resistance develops, limiting clinical benefit. Some mechanisms of resistance have been reported, including secondary FGFR2 kinase domain mutations. Here, we sought to establish the landscape of acquired resistance to FGFR inhibition and to validate findings in model systems.
Experimental design: We examined the spectrum of acquired resistance mechanisms detected in circulating tumor DNA or tumor tissue upon disease progression following FGFR inhibitor therapy in 82 FGFR2-altered cholangiocarcinoma patients from 12 published reports. Functional studies of candidate resistance alterations were performed.
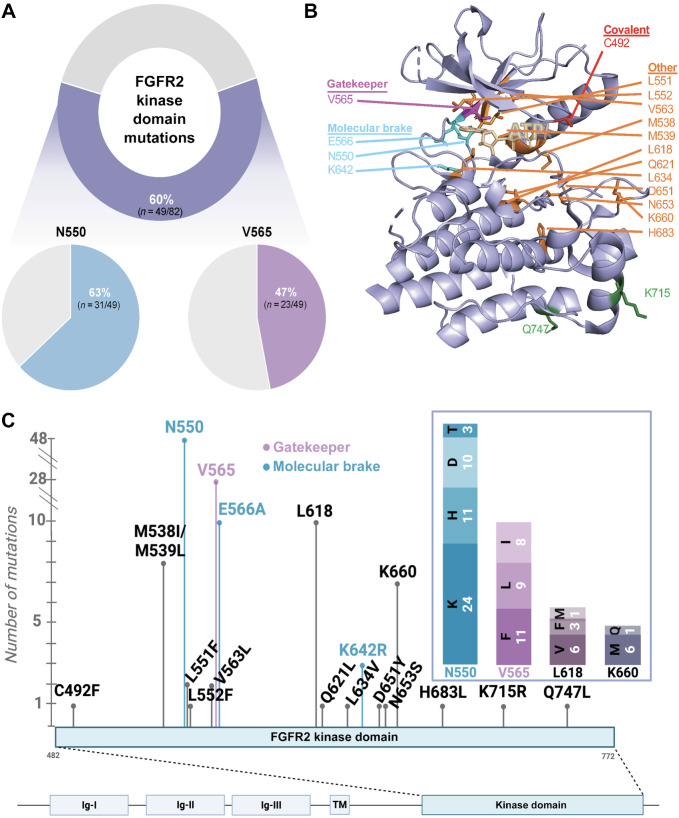
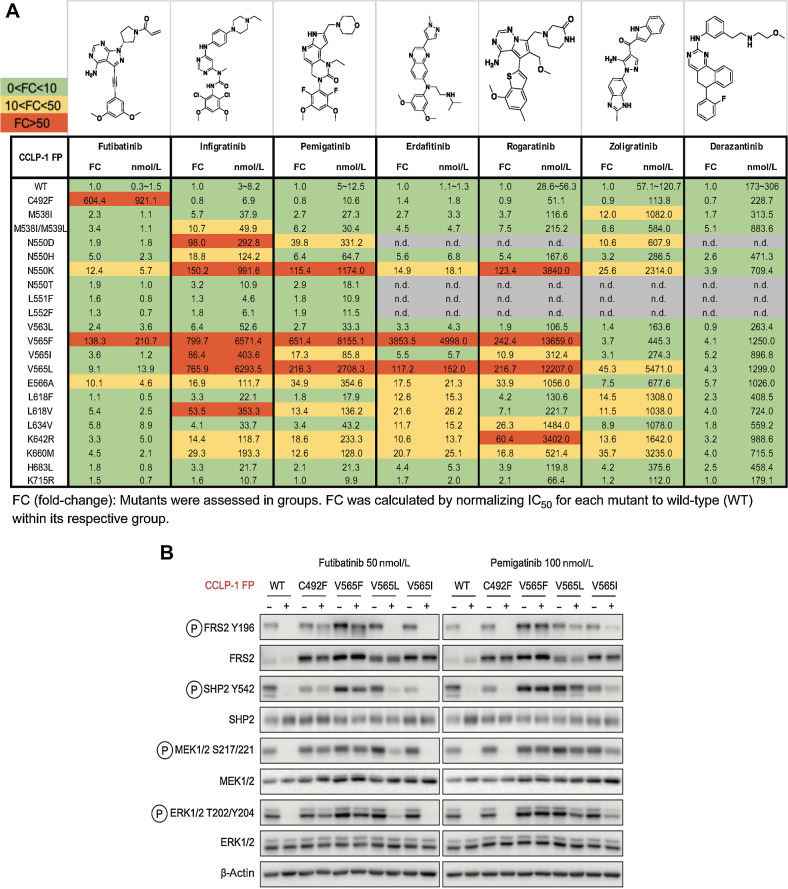
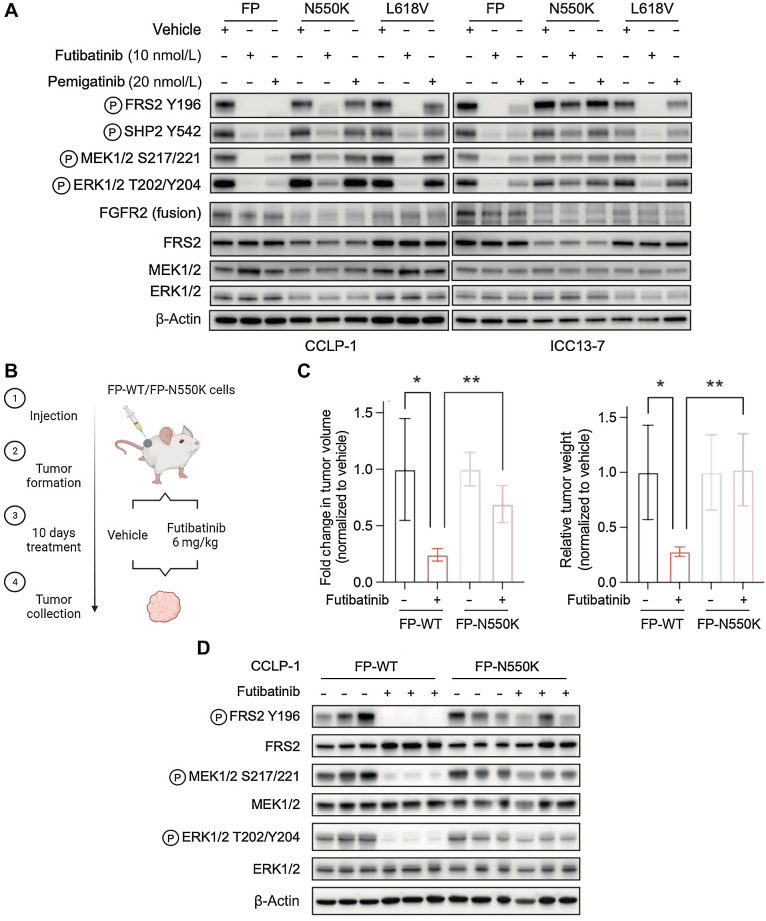
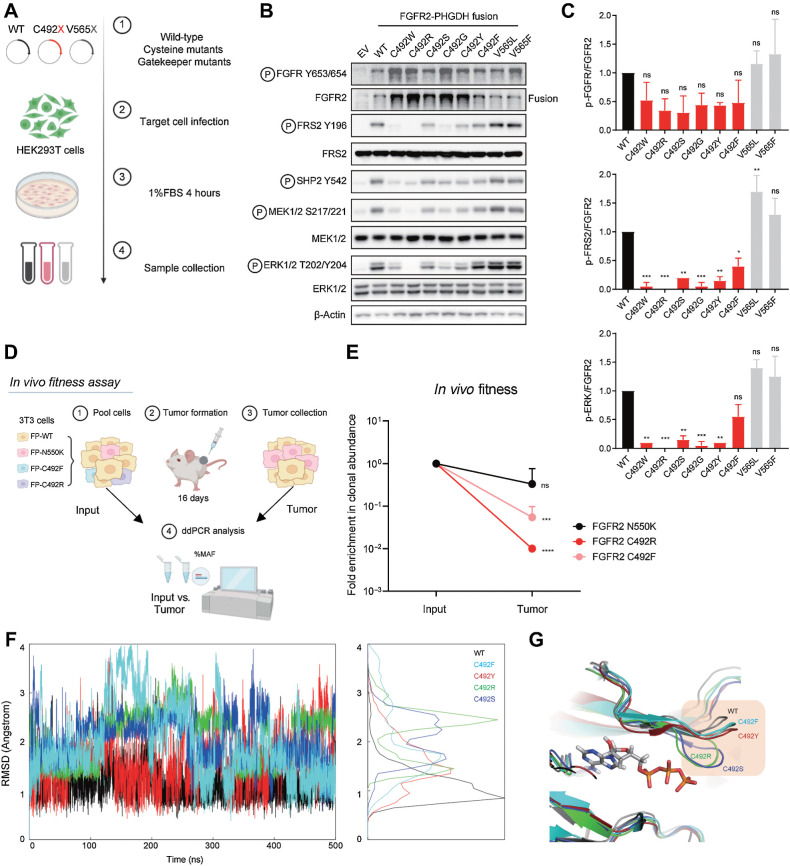
Results: Overall, 49 of 82 patients (60%) had one or more detectable secondary FGFR2 kinase domain mutations upon acquired resistance. N550 molecular brake and V565 gatekeeper mutations were most common, representing 63% and 47% of all FGFR2 kinase domain mutations, respectively. Functional studies showed different inhibitors displayed unique activity profiles against FGFR2 mutations. Interestingly, disruption of the cysteine residue covalently bound by futibatinib (FGFR2 C492) was rare, observed in 1 of 42 patients treated with this drug. FGFR2 C492 mutations were insensitive to inhibition by futibatinib but showed reduced signaling activity, potentially explaining their low frequency.
Conclusions: These data support secondary FGFR2 kinase domain mutations as the primary mode of acquired resistance to FGFR inhibitors, most commonly N550 and V565 mutations. Thus, development of combination strategies and next-generation FGFR inhibitors targeting the full spectrum of FGFR2 resistance mutations will be critical.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, et al. . Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003–10. - PubMed
-
- Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al. . Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427–34. - PubMed
-
- Graham RP, Barr Fritcher EG, Pestova E, Schulz J, Sitailo LA, Vasmatzis G, et al. . Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014;45:1630–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
