

Determinants of Kidney Failure in Primary Hyperoxaluria Type 1: Findings of the European Hyperoxaluria Consortium
- PMID: 37849991
- PMCID: PMC10577369
- DOI: 10.1016/j.ekir.2023.07.025
Determinants of Kidney Failure in Primary Hyperoxaluria Type 1: Findings of the European Hyperoxaluria Consortium
Abstract
Introduction: Primary hyperoxaluria type 1 (PH1) has a highly heterogeneous disease course. Apart from the c.508G>A (p.Gly170Arg) AGXT variant, which imparts a relatively favorable outcome, little is known about determinants of kidney failure. Identifying these is crucial for disease management, especially in this era of new therapies.
Methods: In this retrospective study of 932 patients with PH1 included in the OxalEurope registry, we analyzed genotype-phenotype correlations as well as the impact of nephrocalcinosis, urolithiasis, and urinary oxalate and glycolate excretion on the development of kidney failure, using survival and mixed model analyses.
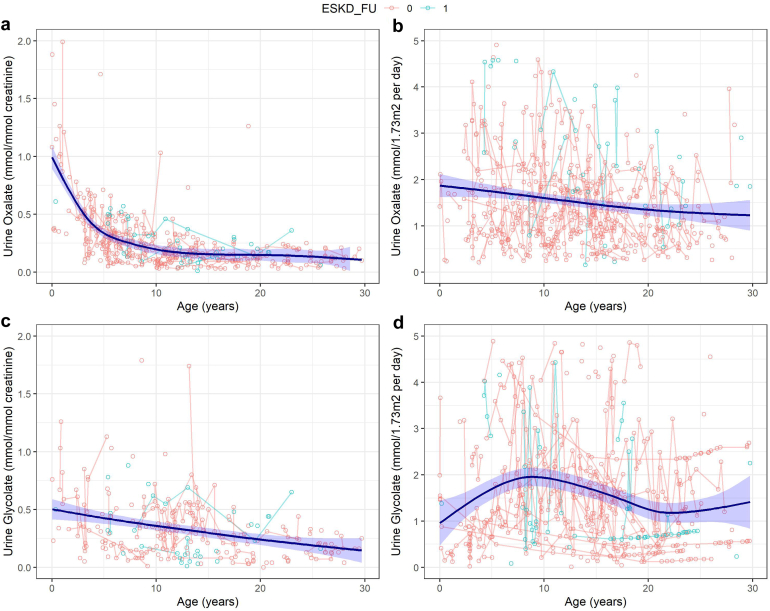
Results: The risk of developing kidney failure was the highest for 175 vitamin-B6 unresponsive ("null") homozygotes and lowest for 155 patients with c.508G>A and c.454T>A (p.Phe152Ile) variants, with a median age of onset of kidney failure of 7.8 and 31.8 years, respectively. Fifty patients with c.731T>C (p.Ile244Thr) homozygote variants had better kidney survival than null homozygotes (P = 0.003). Poor outcomes were found in patients with other potentially vitamin B6-responsive variants. Nephrocalcinosis increased the risk of kidney failure significantly (hazard ratio [HR] 3.17 [2.03-4.94], P < 0.001). Urinary oxalate and glycolate measurements were available in 620 and 579 twenty-four-hour urine collections from 117 and 87 patients, respectively. Urinary oxalate excretion, unlike glycolate, was higher in patients who subsequently developed kidney failure (P = 0.034). However, the 41% intraindividual variation of urinary oxalate resulted in wide confidence intervals.
Conclusion: In conclusion, homozygosity for AGXT null variants and nephrocalcinosis were the strongest determinants for kidney failure in PH1.
Keywords: kidney failure; nephrocalcinosis; primary hyperoxaluria; urinary glycolate; urinary oxalate; urolithiasis.
© 2023 International Society of Nephrology. Published by Elsevier Inc.
Figures






Comment in
-
Is Genotype the Major Outcome Parameter of Kidney Failure in Patients With Primary Hyperoxaluria Type 1?Kidney Int Rep. 2023 Sep 7;8(11):2187-2190. doi: 10.1016/j.ekir.2023.09.012. eCollection 2023 Nov. Kidney Int Rep. 2023. PMID: 38025235 Free PMC article. No abstract available.
References
-
- Almardini R.I., Alfarah M.G., Salaita G.M. The clinical pattern of primary hyperoxaluria in pediatric patient at Queen Rania Abdulla Children Hospital. Arab J Nephrol Transplant. 2014;7:119–123. - PubMed
LinkOut - more resources
Full Text Sources
