

Single-centre experience with autosomal recessive limb-girdle muscular dystrophy: case series and literature review
- PMID: 37852290
- PMCID: PMC10631857
- DOI: 10.1055/s-0043-1772833
Single-centre experience with autosomal recessive limb-girdle muscular dystrophy: case series and literature review
Abstract
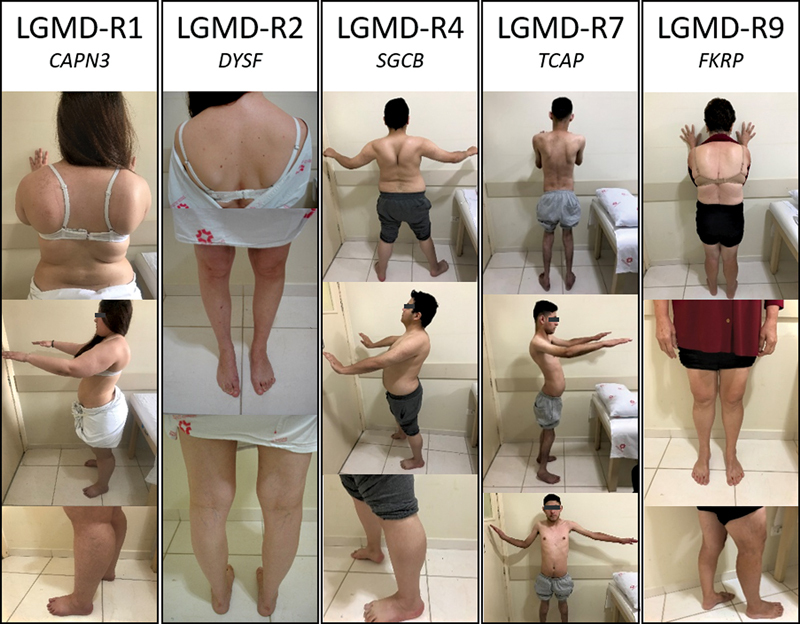
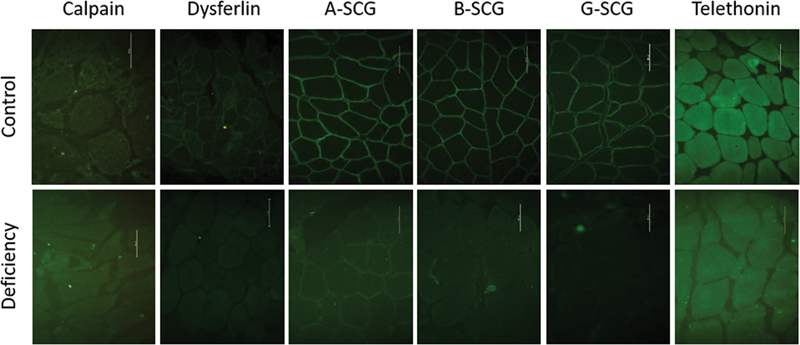
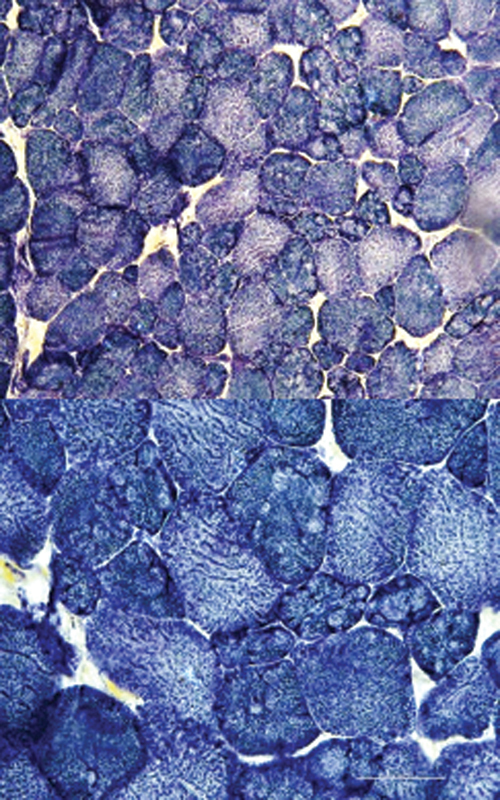
Limb-girdle muscular dystrophy (LGMD) is a group of myopathies that lead to progressive muscle weakness, predominantly involving the shoulder and pelvic girdles; it has a heterogeneous genetic etiology, with variation in the prevalence of subtypes according to the ethnic backgrounds and geographic origins of the populations. The aim of the present study was to analyze a series of patients with autosomal recessive LGMD (LGMD-R) to contribute to a better characterization of the disease and to find the relative proportion of the different subtypes in a Southern Brazil cohort. The sample population consisted of 36 patients with LGMD-R. A 9-gene targeted next-generation sequencing panel revealed variants in 23 patients with LGMD (64%), and it identified calpainopathy (LGMD-R1) in 26%, dysferlinopathy (LGMD-R2) in 26%, sarcoglycanopathies (LGMD-R3-R5) in 13%, telethoninopathy (LGMD-R7) in 18%, dystroglicanopathy (LGMD-R9) in 13%, and anoctaminopathy (LGMD-R12) in 4% of the patients. In these 23 patients with LGMD, there were 27 different disease-related variants in the ANO5, CAPN3, DYSF, FKRP, SGCA, SGCB, SGCG, and TCAP genes. There were different causal variants in different exons of these genes, except for the TCAP gene, for which all patients carried the p.Gln53* variant, and the FKRP gene, which showed recurrence of the p.Leu276Ile variant. We analyzed the phenotypic, genotypic and muscle immunohistochemical features of this Southern Brazilian cohort.
A distrofia muscular de cinturas (DMC) é um grupo de miopatias que leva à fraqueza muscular progressiva, e envolvendo predominante as cinturas escapular e pélvica. A DMCtem uma etiologia genética heterogênea, com variação na prevalência de subtipos de acordo com as origens étnicas e geográficas das populações. O objetivo deste estudo foi analisar uma série de pacientes com DMC do tipo autossômico recessivo (DMC-R) para contribuir para uma melhor caracterização da doença e encontrar a proporção relativa dos diferentes subtipos em uma coorte do Sul do Brasil. A população amostral foi composta por 36 pacientes com DMC-R. O painel de sequenciamento de nova geração com 9 genes revelou variantes em 23 pacientes com DMC (64%), e identificou calpainopatia (DMC-R1) em 26%, disferlinopatia (DMC-R2) em 26%, sarcoglicanopatias (DMC-R3–R5) em 13%, teletoninopatia (D-MCR7) em 18%, distroglicanopatia (D-MCR9) em 13%, e anoctaminopatia (DMC-R12) em 4% dos pacientes. Nesses 23 pacientes com DMC, havia 27 variantes diferentes nos genes ANO5, CAPN3, DYSF, FKRP, SGCA, SGCB, SGCG e TCAP. Foram encontradas diferentes variantes em diferentes éxons desses genes, com exceção do gene TCAP, para o qual todos os pacientes eram portadores da variante p.Gln53*, e do gene FKRP, que apresentou recorrência da variante p.Leu276Ile. As características fenotípicas, genotípicas e imuno-histoquímicas musculares desta coorte do Sul do Brasil foram analisadas.
The Author(s). This is an open access article published by Thieme under the terms of the Creative Commons Attribution 4.0 International License, permitting copying and reproduction so long as the original work is given appropriate credit (https://creativecommons.org/licenses/by/4.0/).
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures
References
-
- Zatz M, de Paula F, Starling A, Vainzof M.The 10 autosomal recessive limb-girdle muscular dystrophies Neuromuscul Disord 200313(7-8):532–544. - PubMed
-
- Khadilkar S V, Patel B A, Lalkaka J A. Making sense of the clinical spectrum of limb girdle muscular dystrophies. Pract Neurol. 2018;18(03):201–210. - PubMed
-
- LGMD workshop study group . Straub V, Murphy A, Udd B. 229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017. Neuromuscul Disord. 2018;28(08):702–710. - PubMed
-
- Liewluck T, Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018;58(02):167–177. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources